L’acromatopsia (ACHM) è una rara malattia retinica ereditaria bilaterale caratterizzata dalla perdita di funzione di tutti e tre i tipi di coni. È anche chiamata «monocromatismo ai bastoncelli» o «cecità totale ai colori»1).
La prevalenza mondiale è stimata in circa 1 persona su 30.0001). La trasmissione è autosomica recessiva, non sono presenti anomalie sistemiche e l’aspettativa di vita è normale.
Esistono due forme di ACHM: completa e incompleta. Nella forma completa vi è assenza totale della funzione dei coni; in quella incompleta almeno un sottotipo di cono conserva una funzione residua, con acuità visiva di circa 20/40–20/120 e fotofobia e nistagmo più lievi2).
Sei geni (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6) sono responsabili e in oltre il 90% dei casi è possibile identificare il gene causale1)2). I soli geni CNGA3 e CNGB3 rappresentano l’80-90% dei casi.
Si noti che le anomalie generali della visione dei colori (discromatopsie congenite) sono dovute a un’anomalia di uno o due tipi di fotopigmenti conici e influenzano solo la visione dei colori. L’ACHM è fondamentalmente diversa perché tutti i coni non funzionano, portando a riduzione dell’acuità visiva, nistagmo e fotofobia.
Esiste un caso noto di effetto fondatore sull’isola di Pingelap (Micronesia). Dopo un tifone nel 1700, la mutazione CNGB3 (p.S435F) si è diffusa tra la popolazione drasticamente ridotta, raggiungendo una prevalenza di circa il 10% e un tasso di portatori di circa il 30%1)2).
QQual è la differenza tra acromatopsia e le anomalie generali della visione dei colori (daltonismo)?
A
Le discromatopsie congenite generali sono dovute a un’anomalia di uno o due tipi di fotopigmenti conici, influenzano solo la visione dei colori e l’acuità visiva è normale. Nell’acromatopsia, tutti e tre i tipi di coni sono non funzionanti, quindi oltre alla perdita della visione dei colori, si verificano riduzione dell’acuità visiva (circa 0,1 o meno), nistagmo, fotofobia ed emeralopia, il che la distingue fondamentalmente.
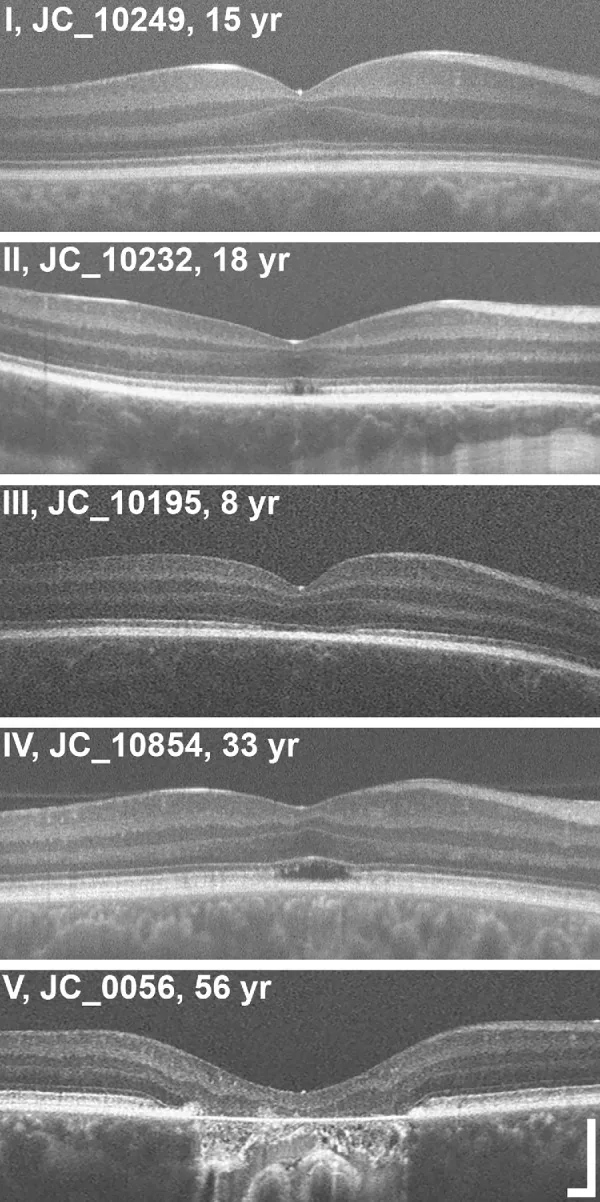
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Classificazione in cinque gradi dell’EZ mediante OCT: EZ normale (I), interruzione dell’EZ (II), scomparsa dell’EZ (III), area a bassa riflettività (IV), atrofia dello strato retinico esterno e RPE (V). Ciò corrisponde alle anomalie della zona ellissoidale trattate nella sezione «2. Principali sintomi e segni clinici».
I sintomi dell’ACHM iniziano a comparire entro le prime settimane di vita.
Riduzione dell’acuità visiva: nella forma completa inferiore a 20/200 (circa 0,1), nella forma incompleta circa 20/80 (circa 0,25).
Fotofobia (sensibilità alla luce): il 38% dei pazienti intervistati l’ha indicata come il sintomo più importante2). In ambienti luminosi, la funzione visiva diminuisce notevolmente.
Emeralopia (cecità diurna): la funzione visiva diminuisce in condizioni di luce intensa, mentre è relativamente buona in ambienti poco illuminati.
Achromatopsia/difetto della visione dei colori: Perdita della visione dei colori su tutti e tre gli assi1). Le tavole di Ishihara sono quasi illeggibili tranne la tavola dimostrativa. Il test Panel D-15 mostra linee di confusione oblique circolari.
Nistagmo pendolare: Compare entro le prime settimane di vita. Tende a migliorare con l’età e diminuisce con la visione da vicino.
Associazione con ipermetropia: L’ipermetropia è comune, ma alcuni presentano un’ampia gamma di errori refrattivi inclusa la miopia.
Risposta pupillare paradossa: Costrizione pupillare iniziale al buio, un reperto caratteristico. Importante indizio per la diagnosi nei bambini.
Reperti del fundus: Inizialmente quasi normale. Anche l’angiografia con fluoresceina mostra reperti quasi normali. Con il tempo possono verificarsi alterazioni a chiazze e atrofia dell’epitelio pigmentato retinico (RPE). Spesso si osserva assenza del riflesso maculare e degenerazione maculare. All’OCT si osservano irregolarità e indistinzione della zona ellissoidale (giunzione segmento interno/esterno dei fotorecettori) e della zona di interdigitazione (estremità dei segmenti esterni dei coni) nello strato esterno della retina.
Reperti elettroretinografici: All’ERG fotopico la risposta dei coni è marcatamente ridotta o assente, mentre la risposta dei bastoncelli all’ERG scotopico è normale o quasi normale1)2). Nel tipo GNAT2 i coni S sono relativamente più preservati rispetto ai tipi CNGA3/CNGB3.
Ipoplasia foveale: Presente nel 60-70% delle mutazioni CNGA3/CNGB32).
FAF (autofluorescenza del fundus): Esistono quattro pattern: normale, aumento del segnale centrale, diminuzione del segnale centrale, anello iperfluorescente + ipofluorescenza centrale2).
Stadiazione OCT: Valutazione delle alterazioni strutturali dello strato esterno della fovea in cinque stadi3).
Mantenimento dello strato esterno della retina (iperriflettività ELM, appiattimento EZ)
Stadio 2
Distruzione della zona ellissoidale (EZ)
Stadio 3
Comparsa di spazio otticamente vuoto
Stadio 4
Spazio otticamente vuoto + distruzione parziale dell’EPR
Stadio 5
Scomparsa dello strato nucleare esterno e/o distruzione completa dell’EPR
In uno studio di follow-up di 10 anni, nonostante la stabilità della migliore acuità visiva corretta (20/400–20/200), è stata confermata una progressione strutturale all’OCT (espansione dello spazio otticamente vuoto: occhio destro 246×59 μm, occhio sinistro 326×53 μm)3).
I foci iperriflettenti compaiono più di 3 anni prima dei cambiamenti dell’EZ, suggerendo che potrebbero essere un marker precoce di progressione della malattia3).
AOSLO (oftalmoscopia laser a scansione con ottica adattiva): Il mosaico dei coni mostra spazi scuri, aumento della distanza inter-coni e ridotta densità dei coni. Non c’è differenza significativa tra i tipi CNGA3 e CNGB3, mentre il tipo GNAT2 mostra una relativa conservazione dei coni2).
Test di visione dei colori: Le tavole di Ishihara sono quasi illeggibili tranne la tavola di dimostrazione. Il Panel D-15 mostra un pattern di errori sull’asse scotopico (tra deutan e tritan). L’anomaloscopio mostra una pendenza ripida e non include l’area di eguagliamento normale.
QL'acuità visiva nell'acromatopsia peggiora con l'età?
A
La migliore acuità visiva corretta spesso rimane stabile a lungo termine. D’altra parte, la valutazione strutturale con OCT può mostrare una progressione nel tempo (distruzione della zona ellissoidale, espansione dello spazio otticamente vuoto)3). È caratteristica una dissociazione tra funzione e struttura, ed è importante un monitoraggio regolare tramite esami.
La trasmissione è autosomica recessiva. Se entrambi i genitori sono portatori, il rischio per il figlio di sviluppare la malattia è del 25%1). La malattia si manifesta spesso in figli di genitori apparentemente sani, e spesso non c’è storia familiare.
È stato riportato anche un modello di ereditarietà non mendeliana dovuto a disomia uniparentale (UPD) paterna. In un caso in cui un omozigote per CNGA3 c.778G>C (p.D260H) è stato stabilito tramite UPD, non è stata rilevata alcuna mutazione nella madre 6). Tali esempi hanno implicazioni importanti per la valutazione del rischio di ricorrenza nella consulenza genetica.
Pattern di mutazione: Prevalentemente mutazioni missenso. Il dominio transmembrana S4 è un hot spot.
Distribuzione geografica: In Medio Oriente e Cina, CNGA3 rappresenta oltre l’80%.
CNGB3
Cromosoma: 8q21.3
Funzione: Subunità beta del canale CNG
Frequenza: Circa il 50% dei casi 1)2)
Pattern di mutazione: Prevalentemente mutazioni nonsenso, frameshift e di splicing. c.1148delC è la mutazione più frequente.
Distribuzione geografica: In Europa e America, CNGB3 rappresenta oltre il 50%.
Altri geni
GNAT2 (1p13.3): trasducina α dei coni. Circa 2%. Forma relativamente lieve con conservazione dello strato dei fotorecettori2)
PDE6C (10q23.33): subunità α della PDE dei coni. Forma grave a esordio precoce5)
PDE6H (12p12.3): subunità γ della PDE dei coni. Estremamente rara1)
ATF6 (1q23.3): fattore di trascrizione della risposta allo stress del reticolo endoplasmatico. Circa 2%. Meccanismo non direttamente coinvolto nella fototrasduzione1)2)
Allele a bassa penetranza: CNGB3 c.1208G>A (p.R403Q) mantiene una funzione parziale, portando a un fenotipo lieve2).
Caratteristiche delle mutazioni PDE6C: In 4 casi con nuove mutazioni (c.1670G>A, c.2192G>A), la triade nistagmo, fotofobia e alterazione della visione dei colori era presente in tutti. L’elettroretinogramma ha mostrato scomparsa della risposta fotopica e del flicker a 30 Hz con risposta scotopica normale, e gli eterozigoti composti presentavano un fenotipo più grave5).
Ereditarietà digenica: Esistono rari casi con mutazioni sia in CNGA3 che in CNGB31).
QPerché un bambino può ammalarsi anche se entrambi i genitori non sono acromati?
A
Poiché l’acromatopsia è autosomica recessiva, anche se ciascun genitore è portatore di una copia del gene mutato, essi sono apparentemente sani e hanno una visione dei colori normale. In una coppia di portatori, c’è una probabilità del 25% che il bambino erediti due copie della mutazione e si ammali1).
In presenza di nistagmo, fotofobia e riduzione dell’acuità visiva nelle prime settimane di vita, è necessaria una valutazione multidisciplinare orientata all’ACHM. La diagnosi definitiva richiede il test genetico.
Elettroretinogramma
Significato diagnostico: Gold standard
Risultati della forma completa: La risposta dei coni all’elettroretinogramma fotopico è assente o gravemente ridotta. La risposta dei bastoncelli all’elettroretinogramma scotopico è normale o quasi normale1)2)
Risultati della forma incompleta: Viene rilevata una debole risposta dei coni corrispondente alla funzione residua dei coni
Caratteristiche del tipo GNAT2: I coni S sono relativamente più preservati rispetto al tipo CNGA3/CNGB3
OCT
Significato diagnostico: Standard per la valutazione strutturale
Contenuto della valutazione: Stadiazione a 5 livelli degli strati esterni della fovea, rilevamento dell’ipoplasia foveale2)3)
Follow-up: Anche in caso di stabilità funzionale, può verificarsi una progressione strutturale, pertanto è importante un monitoraggio regolare
Esami complementari: Valutazione multimodale combinando FAF e AOSLO
Test genetici
Significato diagnostico: Indispensabile per una diagnosi definitiva
Metodo: Pannello mirato di 6 geni (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Tasso di risoluzione: Identificazione del gene causale possibile in oltre il 90% dei casi1)
Partecipazione a studi clinici: La diagnosi molecolare è obbligatoria per partecipare a studi clinici di terapia genica
L’ipermetropia è comune, ma possono essere presenti vari errori refrattivi. La correzione con occhiali o lenti a contatto è importante per massimizzare l’acuità visiva. In caso di ambliopia associata, considerare l’occlusione o la terapia con atropina.
Le lenti colorate per ridurre la fotofobia migliorano significativamente la qualità della vita. In un sondaggio, il 96% dei pazienti preferiva i filtri grigi a quelli rossi, e all’aperto il 74% preferiva i filtri grigi2). È importante una scelta personalizzata in base alle preferenze e all’ambiente di attività del paziente.
È necessaria una spiegazione e un supporto da parte di specialisti riguardo alle caratteristiche dell’ereditarietà autosomica recessiva, al rischio di ricorrenza e al significato della diagnosi genetica. Si raccomanda inoltre di sottoporsi a una diagnosi genetica in preparazione alla partecipazione a futuri studi clinici di terapia genica.
QQuale colore di occhiali filtranti è adatto?
A
In un sondaggio tra i pazienti, il 96% ha preferito i filtri grigi a quelli rossi, e all’aperto il 74% ha preferito i filtri grigi 2). Tuttavia, esistono differenze individuali, quindi è consigliabile provare diversi filtri e scegliere quello più adatto al proprio ambiente di attività dopo aver consultato un oculista o uno specialista in ipovisione.
La cascata di fototrasduzione nei coni normali è la seguente 1).
Buio: Con un’alta concentrazione intracellulare di cGMP, i canali CNG sono aperti, Na⁺ e Ca²⁺ entrano, la cellula rimane depolarizzata e rilascia continuamente glutammato.
Esposizione alla luce: L’opsina (fotopigmento) viene attivata → la trasducina (proteina G) viene attivata → la fosfodiesterasi (PDE) viene attivata → il cGMP viene degradato → i canali CNG si chiudono → iperpolarizzazione → inibizione del rilascio di glutammato.
Feedback negativo: GCAP (proteina attivatrice della guanilato ciclasi) lega il Ca²⁺ e inibisce l’attività di retGC, regolando la produzione di cGMP.
Mutazione CNGA3: Le mutazioni missenso compromettono il ripiegamento, il trasporto intracellulare e l’integrazione di membrana della proteina 1)7). Il dominio transmembrana S4 è un hot spot di mutazione. Sono state riportate oltre 150 mutazioni missenso, di cui 103 con patogenicità non determinata, ma l’analisi struttura-funzione 3D suggerisce che l’86,4% ha conseguenze funzionali simili a mutazioni patogene note 7).
Mutazione CNGB3: Mutazioni nonsenso e frameshift producono proteine del canale troncate o non funzionali 1). In assenza di CNGB3, i canali omomerici CNGA3 persistono, il che può preservare una funzione conica residua.
Mutazione ATF6: Fattore di trascrizione coinvolto nella risposta alle proteine mal ripiegate del reticolo endoplasmatico (UPR), non direttamente coinvolto nella cascata di fototrasduzione 1)2). Il meccanismo patogenetico differisce dagli altri geni ed è ancora in fase di studio.
Il canale CNG ha una struttura tetramerica (CNGA3 × 3 e CNGB3 × 1, secondo alcuni rapporti 2:2), ogni subunità possiede sei domini transmembrana, un dominio di legame dei nucleotidi ciclici, una regione C-linker e un dominio che forma il poro 1).
Meccanismo di degenerazione dei coni e progressione
Lo sviluppo e la morfologia postnatale dei coni sono quasi normali e si ritiene che la degenerazione inizi nella giovane età adulta. L’accumulo di cGMP è coinvolto nel processo degenerativo e una progressione più rapida nelle aree ricche di coni S è stata dimostrata in modelli animali 1).
Tradizionalmente, l’ACHM era considerata una malattia non progressiva. Tuttavia, l’osservazione a lungo termine tramite OCT ha mostrato che, sebbene la migliore acuità visiva corretta rimanga quasi stabile, i cambiamenti strutturali (rottura della zona ellissoidale, allargamento degli spazi ottici) progrediscono 3). Questa dissociazione funzione-struttura è importante per valutare la finestra terapeutica della terapia genica.
Nella zona senza bastoncelli della fovea (area densa di coni priva di bastoncelli), la perdita di coni è marcata, mentre nella regione parafoveale i bastoncelli contribuiscono alla banda EZ, compensando la funzione 3).
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Lo studio NCT03001310 (AAV8-hCARp.hCNGB3) su 23 soggetti (11 adulti e 12 bambini) ha mostrato una sicurezza accettabile. Un miglioramento della visione dei colori è stato osservato in 6/23, un miglioramento della fotofobia in 11/20 e un miglioramento della qualità della vita (QoL) in 21/23. A dosi elevate è stata osservata anche una tendenza all’aumento dell’infiammazione intraoculare2).
Nello studio AAV8.CNGA3 (RD-CURE), 9 pazienti hanno ricevuto 3 dosi (1×10¹⁰–1×10¹¹ vg/occhio). I dati a 1 e 3 anni hanno mostrato una tendenza al miglioramento dell’acuità visiva e della sensibilità al contrasto, senza raggiungere la significatività statistica. La sicurezza era buona1)2).
McKyton et al. (2021) hanno eseguito una mappatura visiva corticale tramite fMRI dopo iniezione sottoretinica di AAV2tYF-PR1.7-hCNGA3 (NCT02935517) in due adulti con ACHM da CNGA34). L’occhio trattato ha mostrato una tolleranza alla luce 5 volte superiore (miglioramento drammatico della fotofobia), acquisizione della capacità di rilevamento del rosso e riduzione delle dimensioni del campo recettivo di popolazione (pRF) (che suggerisce un miglioramento della risoluzione spaziale). D’altra parte, non è stata osservata attivazione delle aree corticali specifiche per la visione dei colori (come V4) e l’elettroretinogramma a campo pieno non ha rilevato risposte dei coni. I pazienti hanno riportato miglioramenti nella vita quotidiana, come “maggiore senso di sicurezza agli attraversamenti pedonali”, “nessun bisogno di lente d’ingrandimento” e “nessun bisogno di occhiali da sole all’aperto”.
Diversi modelli animali hanno confermato il recupero funzionale dopo la sostituzione genica1)2).
Modello murino: Recupero fino all’80% del normale all’elettroretinogramma.
Modello canino (CNGB3): Recupero dell’elettroretinogramma a sfarfallio dei coni dopo 2,5 anni di follow-up post-sostituzione genica. Miglioramento comportamentale in ambienti con 25 lux o più.
Modello ovino (CNGA3): Miglioramento a lungo termine per almeno 6 anni dopo la sostituzione genica.
Primati non umani (PDE6C): Recupero funzionale confermato.
Per quanto riguarda la finestra terapeutica efficace, gli studi sugli animali hanno dimostrato che il trattamento in giovane età è più efficace. I topi anziani hanno mostrato una risposta scarsa, ma il pretrattamento con CNTF (fattore neurotrofico ciliare) ha permesso il recupero funzionale anche nei cani anziani1)2).
È in corso uno studio clinico (NCT04041232) sul fenilbutirrato di glicerolo (PBA) che mira alla risposta allo stress del reticolo endoplasmatico2). Questo approccio mira ad alleviare il misfolding del reticolo endoplasmatico piuttosto che sostituire il gene.
Le principali sfide della terapia genica sono le seguenti1)2)4).
Immunogenicità: La risposta immunitaria al capside AAV può limitare l’efficacia terapeutica.
Ottimizzazione del dosaggio: Necessità di bilanciare l’efficacia terapeutica e il rischio di infiammazione intraoculare.
Sfide dello sviluppo: Difficoltà dell’intervento chirurgico nei bambini e gestione dell’ambliopia.
Limiti della plasticità della corteccia visiva: Negli adulti la riorganizzazione corticale può essere difficile, quindi il momento del trattamento è cruciale.
Efficacia a lungo termine e costi: Garanzia di un effetto duraturo e sfide medico-economiche.
QLa terapia genica può curare completamente l'acromatopsia?
A
Attualmente siamo nella fase di studi di fase I/II di sicurezza ed efficacia. Sono state riportate una riduzione della fotofobia e un certo miglioramento della sensibilità alla luce, ma non è stato raggiunto un completo recupero della visione dei colori2)4). Studi sugli animali hanno dimostrato che il trattamento in giovane età potrebbe essere efficace, ma i dati a lungo termine sull’uomo sono ancora limitati. È importante monitorare i progressi della ricerca e aggiornare regolarmente le informazioni con il medico curante.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.