Achromatopsia (ACHM) is a rare bilateral inherited retinal disease in which all three types of cone photoreceptors lose function. It is also called rod monochromatism or total color blindness 1).
The worldwide prevalence is estimated at approximately 1 in 30,000 people 1). It follows an autosomal recessive inheritance pattern, is not associated with systemic abnormalities, and life expectancy is normal.
ACHM has two forms: complete and incomplete. In the complete form, all cone function is absent; in the incomplete form, at least one cone subtype retains some function, with visual acuity around 20/40 to 20/120 and milder photophobia and nystagmus2).
Six causative genes have been identified (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), and the causative gene can be identified in over 90% of cases 1)2). CNGA3 and CNGB3 alone account for 80-90% of all cases.
Note that common color vision deficiencies (congenital color vision abnormalities) are caused by abnormalities in one or two types of cone photopigments and affect only color vision. ACHM is fundamentally different in that all cones are nonfunctional, accompanied by reduced visual acuity, nystagmus, and photophobia.
There is a known case of founder effect on Pingelap Atoll (Micronesia). After a typhoon in the 1700s drastically reduced the population, the CNGB3 mutation (p.S435F) spread among the islanders, reaching a prevalence of about 10% and a carrier rate of about 30%1)2).
QHow is achromatopsia different from common color vision deficiency (color weakness)?
A
Common congenital color vision abnormalities are caused by abnormalities in one or two types of cone photopigments, affecting only color vision, with normal visual acuity. Achromatopsia involves loss of function of all three cone types, so in addition to color vision loss, it is fundamentally different in that it is accompanied by reduced visual acuity (about 0.1 or less), nystagmus, photophobia, and hemeralopia.
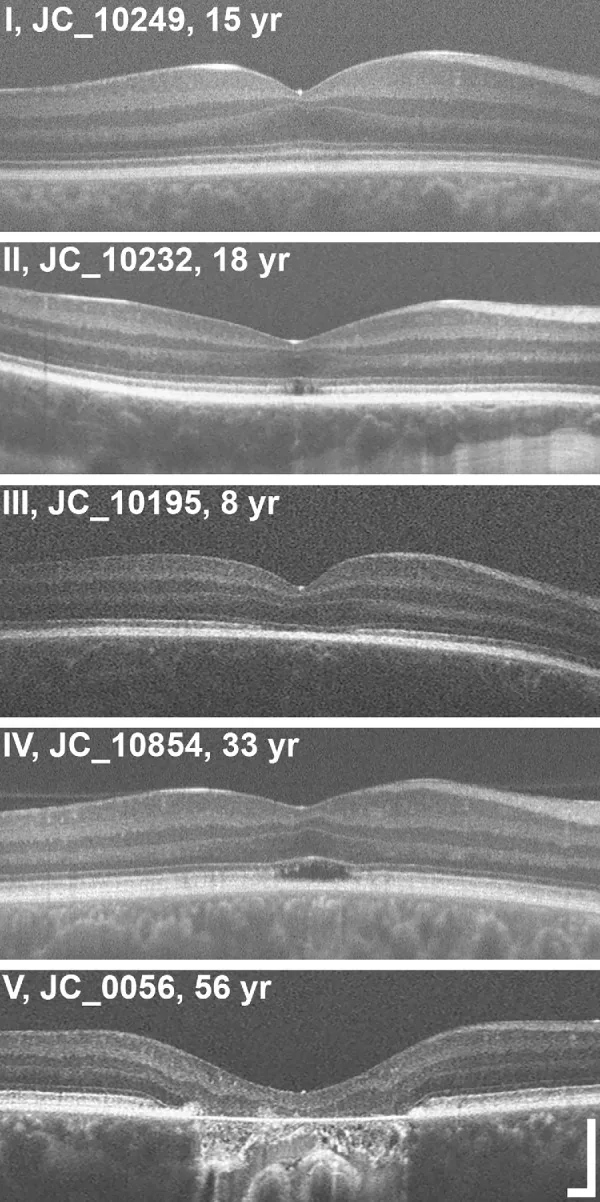
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Five-grade classification of EZ on OCT, showing normal EZ (I), disruption of EZ (II), loss of EZ (III), hyporeflective zone (IV), and outer retinal atrophy with RPE (V). This corresponds to the ellipsoid zone abnormalities discussed in section “2. Main symptoms and clinical findings.”
Symptoms of ACHM begin to appear within a few weeks after birth.
Reduced visual acuity: In complete type, 20/200 (about 0.1) or less; in incomplete type, about 20/80 (about 0.25).
Photophobia (light sensitivity): 38% of patients in a survey reported it as the most significant symptom2). Visual function markedly decreases in bright environments.
Hemeralopia (day blindness): Visual function decreases in bright light, while relatively good vision is maintained in dim environments.
Color vision deficiency/loss: Complete loss of color vision in all three axes1). Ishihara color plates are nearly unreadable except for the demonstration plate. Panel D-15 test shows circular oblique confusion lines.
Nystagmus (pendular nystagmus): Appears within weeks after birth. Tends to improve with age and decreases during near vision.
Associated hyperopia: Hyperopia is common, but a wide range of refractive errors including myopia may also occur.
Paradoxical pupil response: Characteristic finding of initial pupillary constriction in darkness. An important clue for diagnosis in children.
Fundus findings: Initially appears nearly normal. Fluorescein angiography also shows almost normal findings. Over time, patchy changes and atrophy of the retinal pigment epithelium (RPE) may occur. Absence of foveal reflex and macular degeneration are often observed. OCT shows irregular and indistinct ellipsoid zone and interdigitation zone of the outer retina.
Electroretinogram findings: Photopic ERG shows markedly reduced or absent cone responses, while scotopic ERG shows normal to nearly normal rod responses1)2). In GNAT2 type, S-cones are relatively preserved compared to CNGA3/CNGB3 type.
Foveal hypoplasia: Present in 60–70% of cases with CNGA3/CNGB3 mutations2).
FAF (Fundus Autofluorescence): Four patterns exist: normal, increased central signal, decreased central signal, and hyperfluorescent ring with central hypofluorescence2).
OCT staging: Structural changes in the outer foveal layers are evaluated in 5 stages3).
Preservation of outer retinal layers (ELM hyperreflectivity, EZ flattening)
Stage 2
Disruption of the ellipsoid zone (EZ)
Stage 3
Appearance of optically empty space
Stage 4
Optically empty space + partial RPE disruption
Stage 5
Loss of outer nuclear layer and/or complete RPE disruption
In a 10-year follow-up study, despite stable best-corrected visual acuity (20/400 to 20/200), structural progression on OCT (expansion of optically empty space: right eye 246×59 μm, left eye 326×53 μm) was confirmed3).
Hyperreflective foci appear more than 3 years before changes in the ellipsoid zone, suggesting they may be early markers of disease progression3).
AOSLO (Adaptive Optics Scanning Laser Ophthalmoscopy): The cone mosaic shows dark spaces, increased cone spacing, and decreased cone density. No significant difference between CNGA3 and CNGB3 types; GNAT2 type shows relatively preserved cones2).
Color vision testing: Ishihara color plates are almost unreadable except for the demonstration plate. Panel D-15 shows a scotopic axis error pattern (between deutan and tritan). The anomaloscope shows a steep slope and does not include the normal matching range.
QDoes visual acuity in achromatopsia worsen with age?
A
Best-corrected visual acuity is often stable over the long term. However, structural evaluation by OCT may show progressive changes (disruption of the ellipsoid zone, expansion of optically empty space) over time3). A dissociation between function and structure is characteristic, and regular monitoring is important.
Autosomal recessive inheritance. If both parents are carriers, the risk of an affected child is 25%1). Affected individuals often have unaffected parents, and a family history may be absent.
Non-Mendelian inheritance patterns due to paternal uniparental disomy (UPD) have also been reported. In a case where homozygosity for CNGA3 c.778G>C (p.D260H) resulted from UPD, no mutation was detected in the mother 6). Such examples have important implications for recurrence risk assessment in genetic counseling.
ATF6 (1q23.3): Endoplasmic reticulum stress response transcription factor. Approximately 2%. Mechanism not directly involved in phototransduction 1)2)
Hypomorphic allele: CNGB3 c.1208G>A (p.R403Q) retains partial function, resulting in a mild phenotype 2).
Characteristics of PDE6C mutations: In four cases with novel mutations (c.1670G>A, c.2192G>A), the triad of nystagmus, photophobia, and color vision defects was observed in all. Electroretinography showed absent photopic and 30 Hz flicker responses with normal scotopic responses, and compound heterozygotes exhibited a more severe phenotype 5).
Digenic inheritance: Rare cases with mutations in both CNGA3 and CNGB3 exist 1).
QWhy can a child develop achromatopsia even if both parents do not have it?
A
Because achromatopsia is autosomal recessive, both parents may be carriers (each with one copy of the mutated gene) and appear healthy with normal color vision. When two carriers have a child, there is a 25% chance that the child inherits two copies of the mutation and develops the condition 1).
If nystagmus, photophobia, and reduced visual acuity are observed within the first few weeks of life, comprehensive testing with ACHM in mind is necessary. Genetic testing is essential for a definitive diagnosis.
Electroretinography
Diagnostic significance: Gold standard
Complete type findings: Photopic electroretinogram shows absent or severely reduced cone responses. Scotopic electroretinogram shows normal to nearly normal rod responses1)2)
Incomplete type findings: Weak cone responses corresponding to residual cone function are detected
GNAT2 type characteristics: S cones are relatively preserved compared to CNGA3/CNGB3 type
OCT
Diagnostic significance: Standard for structural evaluation
Evaluation content: 5-stage grading of the outer foveal layers, detection of foveal hypoplasia2)3)
Follow-up: Even if functionally stable, structural progression can occur, so regular monitoring is important
Auxiliary tests: Multimodal evaluation combining FAF and AOSLO
Genetic testing
Diagnostic significance: Essential for definitive diagnosis
Method: Targeted panel of 6 genes (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Resolution rate: Causative gene can be identified in over 90% of cases1)
Clinical trial participation: Molecular diagnosis is mandatory for participation in gene therapy clinical trials
Hyperopia is common, but a wide range of refractive errors can occur. Correction with glasses or contact lenses is important to maximize visual acuity. If amblyopia is present, occlusion therapy or atropine treatment may be considered.
Tinted lenses to reduce photophobia greatly improve quality of life. Patient surveys show that 96% prefer gray filters over red filters, and 74% prefer gray filters outdoors2). Selection should be tailored to individual patient preferences and activity environment.
Expert explanation and support are needed regarding the characteristics of autosomal recessive inheritance, recurrence risk, and the significance of genetic diagnosis. It is also recommended to undergo genetic testing in preparation for future participation in gene therapy clinical trials.
QWhat color of light-filtering glasses is suitable?
A
In a patient survey, 96% preferred gray filters over red filters, and 74% preferred gray filters outdoors2). However, since individual differences exist, it is advisable to try various filters and choose one that suits your activity environment in consultation with an ophthalmologist or low vision specialist.
The phototransduction cascade in normal cone photoreceptors is as follows1).
Dark state: Intracellular cGMP concentration is high, CNG channels are open, Na⁺ and Ca²⁺ influx maintains depolarization, and glutamate is continuously released.
Light exposure: Opsin (visual pigment) activates → transducin (G protein) activates → phosphodiesterase (PDE) activates → cGMP is hydrolyzed → CNG channels close → hyperpolarization → glutamate release is suppressed.
CNGA3 mutations: Missense mutations impair protein folding, intracellular trafficking, and membrane integration1)7). The S4 transmembrane domain is a mutational hotspot. Over 150 missense mutations have been reported; 103 were of uncertain pathogenicity, but 3D structure-function analysis suggests 86.4% have functional consequences similar to known pathogenic mutations7).
CNGB3 mutations: Nonsense and frameshift mutations produce truncated or loss-of-function channel proteins1). In CNGB3 deficiency, residual CNGA3 homomeric channels may preserve some cone function.
ATF6 mutations: ATF6 is a transcription factor involved in the unfolded protein response (UPR) of the endoplasmic reticulum and does not directly participate in the phototransduction cascade1)2). Its pathogenic mechanism differs from other genes and is still under investigation.
The CNG channel has a tetrameric structure (CNGA3 × 3 and CNGB3 × 1, or 2:2 in some reports), with each subunit containing six transmembrane domains, a cyclic nucleotide-binding domain, a C-linker region, and a pore-forming domain1).
Postnatal cone development and morphology are nearly normal, and degeneration is thought to begin in young adulthood. cGMP accumulation is involved in the degenerative process, and animal models show more rapid progression in S-cone-rich regions1).
Traditionally, ACHM was considered a non-progressive disease. However, long-term OCT observation has demonstrated that structural changes (EZ disruption, enlargement of optical gaps) progress despite nearly stable best-corrected visual acuity3). This functional-structural dissociation has important implications for evaluating the therapeutic window for gene therapy.
In the foveal rod-free zone (cone-dense area without rods), cone loss is prominent, whereas in the parafovea, rods contribute to the EZ band, compensating for function 3).
7. Latest Research and Future Perspectives (Investigational Reports)
In a trial of NCT03001310 (AAV8-hCARp.hCNGB3) involving 11 adults and 12 children (total 23 participants), safety was within acceptable range. Color vision improvement was observed in 6/23, photophobia improvement in 11/20, and quality of life (QoL) improvement in 21/23. A trend of increased intraocular inflammation was also observed at high doses 2).
In the AAV8.CNGA3 (RD-CURE) trial, 9 participants received three doses (1×10¹⁰ to 1×10¹¹ vg/eye). At 1-year and 3-year follow-ups, trends of improvement in visual acuity and contrast sensitivity were observed, but did not reach statistical significance. Safety was good 1)2).
McKyton et al. (2021) performed fMRI cortical visual mapping after subretinal injection of AAV2tYF-PR1.7-hCNGA3 (NCT02935517) in two adults with CNGA3-ACHM 4). The treated eye showed 5-fold greater light tolerance (dramatic improvement in photophobia) compared to the untreated eye, acquisition of red detection ability, and reduction in population receptive field (pRF) size (suggesting improved spatial resolution). However, no activation of color-specific cortical areas (e.g., V4) was observed, and cone responses remained undetectable on full-field electroretinography. Patients self-reported improvements in daily life such as “increased safety at crosswalks,” “no need for magnifiers,” and “no need for sunglasses outdoors.”
Dog model (CNGB3): Cone flicker electroretinography recovery at 2.5-year follow-up after gene supplementation. Behavioral improvement in environments above 25 lux.
Sheep model (CNGA3): Long-term improvement for at least 6 years after gene supplementation.
Regarding the therapeutic time window, animal studies have shown that treatment at a younger age is more effective. Older mice showed poor response, but pretreatment with CNTF (ciliary neurotrophic factor) enabled functional recovery even in older dogs 1)2).
A clinical trial (NCT04041232) of phenylbutyrate glycerol (PBA) targeting the endoplasmic reticulum stress response is ongoing 2). This approach is attracting attention as a method to alleviate misfolding in the endoplasmic reticulum rather than gene supplementation.
The main challenges of gene therapy are as follows 1)2)4).
Immunogenicity: Immune response to the AAV capsid may limit therapeutic efficacy.
Dosage optimization: Balancing therapeutic effect and risk of intraocular inflammation is necessary.
Developmental challenges: Difficulty of surgery in pediatric eyes and management of amblyopia.
Limitations of visual cortex plasticity: Cortical functional reorganization may be difficult in adults, making treatment timing important.
Long-term efficacy and cost: Ensuring sustained effect and health economic challenges.
QCan gene therapy completely cure achromatopsia?
A
Currently, it is at the phase I/II safety and efficacy trial stage. Reduction of photophobia and some improvement in light sensitivity have been reported, but complete color vision restoration has not been achieved2)4). Animal studies suggest that treatment at a young age may be effective, but long-term human data are still limited. It is important to monitor research progress and regularly update information with the attending physician.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.