Retinitis pigmentosa (RP) is a general term for a group of hereditary diseases characterized by progressive, widespread degeneration primarily affecting photoreceptors (rods and cones) and the retinal pigment epithelium (RPE). When rod degeneration precedes cone degeneration, it is called rod-cone dystrophy, and RP is understood synonymously. It is not a single disease but a group of disorders involving more than 100 genes.
The prevalence is 1 in 4,000 to 8,000, and the total number of patients in Japan exceeds at least 30,000 (20,687 recipients of designated intractable disease benefits in fiscal year 2023) 9). Regarding visual impairment, RP is the second leading cause (13.0%) among new physical disability certificate holders aged 18 and over (after glaucoma at 40.7% in fiscal year 2019) and the leading cause of congenital blindness 9). In Japan, it has been designated as an intractable disease under the Intractable Disease Act (since January 1, 2015) 9) and is eligible for medical expense subsidies.
RP also includes syndromic RP accompanied by other systemic diseases, and is classified as follows with ciliopathy as a higher concept 9)2).
Ciliopathy:
Usher syndrome (type 1/2/3): RP + hearing loss; designated intractable disease (AR). Type 1 involves severe hearing loss and vestibular dysfunction from early childhood.
Also, differentiation from various syndromes such as PHARC (polyneuropathy, hearing loss, ataxia, RP, cataract), PCARP, and Oliver-McFarlane syndrome is important 3).
QIs retinitis pigmentosa hereditary?
A
RP is a hereditary disease, but it is not necessarily inherited by everyone. The risk of inheritance to children varies depending on the inheritance pattern. In AD type, there is a 50% chance of inheritance to children, but in AR and XL types, the risk varies according to the inheritance pattern. In sporadic cases (48-63% of all cases), the risk of inheritance to the next generation is often relatively low 9). Use of genetic counseling is recommended.
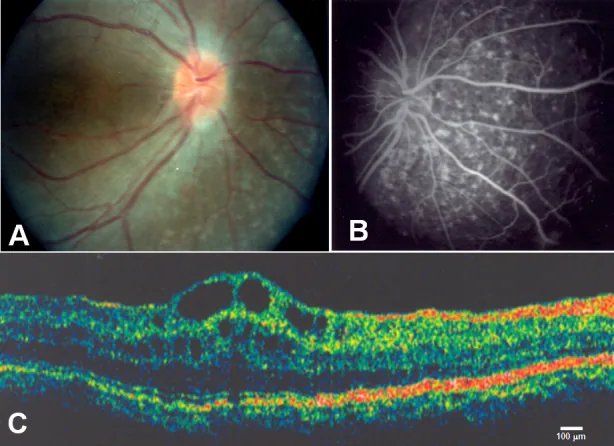
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A shows optic disc drusen and extensive retinal pigment epithelium atrophy, B shows choroidal hyperfluorescence corresponding to retinal pigment epithelium atrophy, C shows cystoid macular edema and separation of the inner retinal layers at the fovea. This corresponds to cystoid macular edema discussed in the section “2. Main symptoms and clinical findings”.
Symptoms of RP change according to the stage of progression. Since rod photoreceptors degenerate first, night blindness appears as the earliest symptom.
Night blindness: Decreased vision or difficulty seeing in dark places. It appears from the earliest stage because rod photoreceptors degenerate first 9); it is noticed as difficulty seeing in dim light in the teens to twenties. In the early stage, daytime visual function is often normal.
Visual field constriction: Gradually narrows from the peripheral visual field inward. Progresses from ring scotoma to concentric visual field constriction (tunnel vision) 9)
Decreased visual acuity: When cone degeneration progresses following rod degeneration, central visual acuity also decreases. When CME is present, moderate visual acuity loss may occur relatively early. In some cases, central visual acuity is preserved until the end stage.
Photophobia (hemeralopia): Sensitivity to light. A manifestation of cone dysfunction. Increases as cone degeneration progresses. Differentiation from light scattering due to cataract is important.
Photopsia: May occur with degeneration and loss of photoreceptors.
Visual hallucinations (Charles Bonnet syndrome): A phenomenon in which patients with advanced vision loss see landscapes or people that do not actually exist. This is not a pathological experience but a phenomenon caused by hyperactivity of the visual cortex 9)
The following shows the approximate progression of symptoms by disease stage.
Stage
Main symptoms
Approximate age
Early
Night blindness
10s–20s
Intermediate
Visual field constriction (ring scotoma → concentric)
30s–40s
Late
Vision loss, color vision abnormalities, photophobia
Posterior subcapsular cataract (PSC): Occurs in about 50% of cases. Characterized by decreased vision in bright light. An EZ (ellipsoid zone) width of 600 μm or more predicts good postoperative visual acuity (AUC 0.97)5).
Cystoid macular edema (CME): Occurs in 10–50% of cases and is a major cause of central vision loss9).
Angle-closure glaucoma: Attacks reported in about 1% of cases; lens subluxation due to weakened zonules may also occur9).
Macular hole / foveoschisis: Relatively rare but may be an indication for vitrectomy9).
QDoes visual acuity improve after cataract surgery?
A
In cataract surgery for posterior subcapsular cataract associated with RP, good postoperative visual acuity can be expected if the preoperative OCT shows an EZ (ellipsoid zone) width of 600 μm or more (AUC 0.97)5). EZ width is a useful biomarker for predicting visual function before surgery. However, zonules are often fragile, requiring caution for anterior capsule contraction and IOL dislocation. For prevention of postoperative CME (10–14%), longer than usual use of steroid and NSAID eye drops is recommended9).
RP is a group of diseases with high genetic heterogeneity caused by mutations in more than 100 genes9). Major causative genes in Japanese patients are shown by inheritance pattern.
A comparison of major causative genes is shown below.
Gene
Inheritance
Frequency/Features in Japanese
EYS
AR
30–50% of cases with identified causative gene (most common in AR type)12)13)
USH2A
AR
Second most common in AR type (4–9%); major gene for Usher syndrome12)
RHO
AD
Most common in AD type6)
RPGR
X-linked
About 70–75% of XL type6)
REEP6
AR
One of the causative genes for AR type4)
The characteristics of each gene are supplemented below.
EYS (Eyes Shut Homolog): The most common causative gene for AR RP in Japanese (30–50% of identified cases)12)13). It is not as frequent in Western populations, reflecting a Japanese-specific genetic background.
USH2A: The main causative gene for Usher syndrome (RP + hearing loss), and the second most common in Japanese AR RP after EYS (4–9%)12)
RHO (Rhodopsin): The most common causative gene for ADRP 6). It encodes the light receptor protein in rod photoreceptors.
RPGR (Retinitis Pigmentosa GTPase Regulator): The main causative gene for XLRP 6). Male patients with RPGR mutations have been reported to develop primary ciliary dyskinesia (PCD) 1).
REEP6 (Receptor Expression-Enhancing Protein 6): One of the causative genes for ARRP 4).
The detection rate of causative genes by genetic testing varies by inheritance pattern. It is reported to be 35–60% for AD, 30–50% for AR and sporadic cases, and 16–36% for XL 6).
In syndromic RP, including Joubert syndrome and Bardet-Biedl syndrome, mutations in cilia-related genes are common and may be accompanied by systemic complications (e.g., renal disease, polydactyly, obesity) 2). It is also important to differentiate from various syndromes such as PHARC, PCARP, and Oliver-McFarlane syndrome 3).
QShould I undergo genetic testing?
A
Genetic diagnosis is important for definitive diagnosis, genetic counseling, and determining eligibility for gene therapy. The PrismGuide IRD Panel System (comprehensive analysis of exon sequences of 82 IRD causative genes) became covered by insurance in 2023, but as of June 2025, it is only indicated for young-onset patients suspected of having RPE65-related IRD 9). It is recommended to be performed in combination with genetic counseling. Genetic counseling can be received without undergoing genetic diagnosis.
Acquired blue-yellow defect is common; Panel D-15 and 100 Hue test9)
Dark adaptation test
Rod function assessment
Kohlrausch break not detected10)
NGS genetic testing
Genetic diagnosis
PrismGuide IRD panel (82 genes) 9)
Details of each test are shown below.
Electroretinography (ERG): Essential for definitive diagnosis 6)9). Rod response (scotopic ERG) decreases from early stages, and cone response (photopic ERG) also decreases with progression. Full-field ERG is standard. Often already non-recordable at the time of examination.
Optical coherence tomography (OCT): Evaluates the width and disappearance pattern of the ellipsoid zone (EZ). EZ width is useful as a quantitative biomarker for visual function and prognosis, and is also used to determine the indication for cataract surgery 5). Thinning of the outer nuclear layer and loss of EZ are observed from early stages.
Fundus autofluorescence (FAF): An abnormal hyperautofluorescent ring (AF ring) appears around the macula, serving as an indicator of disease progression and residual functional retina6).
Visual field testing: Goldmann perimetry (kinetic perimetry) is standard. With progression, ring scotoma leads to concentric visual field constriction 10). Humphrey Field Analyzer (HFA) 10-2 program is useful for evaluating residual central cone function 9).
Dark adaptation testing: The Kohlrausch break (rod-cone transition point) is not detected 10).
Next-generation sequencing (NGS): The PrismGuide IRD panel system can comprehensively analyze exon sequences of 82 causative genes 9). It is also essential for determining eligibility for gene therapy.
Currently, there is no curative treatment for RP 6)9). Treatment focuses on maintaining visual function, managing complications, and supporting social life.
Photoreceptor Protection
Vitamin A (15,000 IU/day): Oral administration has been reported to slow ERG deterioration by a few percent 14). No improvement in visual acuity or visual field. Liver function monitoring is required for long-term use. Contraindicated during pregnancy due to teratogenicity. May accelerate progression in ABCA4 mutations 14). Note that vitamin E may accelerate progression and requires caution 14).
Unoprostone eye drops: Dose-dependent improvement in sensitivity was observed, but the primary endpoint (central 2-degree retinal sensitivity) in Phase 2 trial was not significant 16).
Nilvadipine (calcium channel blocker): Long-term reports suggest slower progression of visual field defects 15). Based on single-center, small-sample reports; multicenter replication has not been performed.
N-acetylcysteine (NAC): Suppresses oxidative stress. Phase I trial reported visual acuity improvement 17); Phase III ongoing as of 2025.
DHA and Lutein: Protect macular photoreceptors from oxidative stress. No additional benefit of DHA when added to vitamin A has been confirmed.
Helenien (Adaptinol): Approved for temporary improvement of visual field and dark adaptation in RP. Efficacy evaluation by modern medical standards has not been performed.
Light-filtering glasses: Reduce oxidative stress from UV and bright light. Daily use is recommended.
First-line treatment is carbonic anhydrase inhibitors (CAI). Use dorzolamide (Trusopt) eye drops or acetazolamide (Diamox) orally. CMT improvement is achieved in about 40%. Recurrence occurs in about 30% 9).
Anti-VEGF drugs are not recommended for RP-CME because VEGF production is decreased 9).
Note that none of these are approved for RP-CME and are used off-label.
Cataract surgery: Performed for cases with posterior subcapsular cataract. Preoperative OCT showing EZ width ≥600 μm is a predictor of good postoperative visual acuity5). In cases with zonular weakness, consider using a capsular tension ring. For postoperative CME (10–14%), use steroid and NSAID eye drops for a longer period than usual 9).
Angle-closure glaucoma: RP patients have a high risk of developing primary angle-closure glaucoma. The anterior chamber gradually becomes shallow; perform prophylactic laser iridotomy or cataract surgery 9).
Epiretinal membrane (GL2026 CQ4): Vitrectomy. Visual improvement can be expected in cases with a continuous EZ line. In cases with a discontinuous EZ line, recovery is limited. Severe macular atrophy has been reported long-term postoperatively; evaluation at a specialized facility is recommended 9).
Macular hole: Vitrectomy is the only curative treatment. Postoperative outcomes have been limitedly studied 9).
Support and Rehabilitation
Low vision care: Low vision → magnifiers, video magnifiers, tablet devices; photophobia → tinted glasses; visual field constriction → white cane; distance vision → monocular telescope; night vision aids. Individualized support according to visual field and acuity is important. Use of Smart Site (introduction to local low vision consultation services) is recommended.
Genetic counseling: Provided by clinical geneticists and certified genetic counselors. Common consultations include recurrence risk estimation, education, employment, marriage, and childbirth. Genetic counseling can be received even without undergoing genetic testing.
Intractable disease system: Medical expense subsidy is available as a designated intractable disease 9). Also consider obtaining a physical disability certificate and Medical Support for Independence.
Voretigene neparvovec (Luxturna): A gene therapy drug that can be administered to patients with biallelic pathogenic variants in the RPE65 gene and sufficient viable retinal cells. Approved in Japan in 2023 9). In the US Phase III (301 trial), 31 patients were enrolled; mITT analysis (20 intervention, 9 control) showed significant improvement in MLMT and white light FST compared to the control group 18). In the domestic Phase III (A11301 trial), FST sensitivity increase and visual field expansion were confirmed in 4 Japanese patients 19). Visual acuity improvement is limited, and long-term complications such as chorioretinal atrophy have been reported in over 20% of patients 9).
Perform the following every 6 months to 1 year: visual acuity, slit-lamp microscopy, fundus examination, Humphrey visual field (HFA 10-2), OCT9).
QWhat drugs are used to treat macular edema?
A
The first-line treatment for RP-CME is carbonic anhydrase inhibitors (CAIs), with dorzolamide eye drops or oral acetazolamide used 9). CMT improvement is achieved in about 40% of cases, but recurrence occurs in about 30%. If CAI is ineffective, intravitreal triamcinolone acetonide injection or dexamethasone intravitreal implant (Ozurdex) are options. Anti-VEGF drugs are not recommended for RP-CME. Note that all of these are used off-label.
The final common pathway of photoreceptor cell death in RP is apoptosis. Although the types of genetic mutations are diverse, they ultimately converge on a common cell death pathway.
In RP, rod photoreceptors typically degenerate and disappear first, followed by secondary degeneration of cone photoreceptors 7). Cones depend on trophic factors produced by rods (RdCVF: Rod-derived Cone Viability Factor) for survival, so after rod loss, cones also lose function 7)11).
The retina is one of the most metabolically active tissues, converting 80–90% of glucose to lactate via aerobic glycolysis (Warburg effect). Cones are more vulnerable to metabolic stress than rods, and this metabolic vulnerability also contributes to secondary cone degeneration 11).
Inflammation is also recognized as a major factor in RP progression, with microglial activation and macrophage infiltration worsening retinal damage 11). Oxidative stress also acts as a biological driver of secondary cone degeneration.
REEP6 mutations: REEP6 encodes a protein involved in ER morphology maintenance. Pathogenic mutations lead to the formation of ER inclusion bodies in the rod outer segment, resulting in photoreceptor degeneration 4)
RPGR mutations: RPGR is involved in the axonemal structure of primary cilia, and mutations impair the transport of substances to the photoreceptor outer segment 1)
7. Latest Research and Future Perspectives (Investigational Reports)
Gene therapy is the most promising approach for treating inherited retinal diseases 8).
Luxturna (voretigene neparvovec): A gene therapy drug for patients with biallelic pathogenic variants in the RPE65 gene. In the US Phase III (301 trial), 31 patients were enrolled, and the mITT analysis (20 intervention, 9 control) showed significant improvement in MLMT and white light FST compared to the control group 18). In the domestic Phase III (A11301 trial), FST sensitivity increase and visual field expansion were confirmed in 4 Japanese patients 19). It was approved in Japan in 2023, serving as a bridge between standard treatment and investigational therapy.
RPGR gene therapy: AAV-mediated gene therapy for XL-RP caused by RPGR mutations has advanced to Phase I/II/III clinical trials 8).
CRISPR/Cas9: Research is ongoing for direct correction of pathogenic mutations and inactivation of dominant negative mutations 8).
RdCVF (Rod-derived Cone Viability Factor) and Cone Protection Therapy
RdCVF is a protein secreted by rods that maintains cone survival 7)11). Clinical trials of cone protection therapy using RdCVF are underway, and it is attracting attention as an independent treatment strategy to preserve cone function after rod degeneration.
NAC is a drug that suppresses oxidative stress, and Phase I trials have reported visual acuity improvement 17). As of 2025, Phase III trials are ongoing.
Potential Repurposing of Glucocorticoids (Dexamethasone)
Recent in vivo studies (rd10 mouse model) have demonstrated that intravitreal dexamethasone protects cone photoreceptors and the retinal pigment epithelium11). Glucocorticoids have strong repurposing potential as mutation-independent therapeutics. However, current evidence is limited to animal models, and further validation is needed for clinical application in humans.
iPS Cell-Derived Retinal Transplantation and Artificial Retina
iPS cell-derived retinal transplantation: Research is progressing on transplanting photoreceptor sheets generated from the patient’s own iPS cells.
Artificial retina (retinal prosthesis): Electrical stimulation devices for end-stage RP. Argus II and others have been commercialized overseas, and clinical trials of the suprachoroidal transretinal stimulation method are ongoing in Japan.
QIs gene therapy available in Japan?
A
Voretigene neparvovec (Luxturna) was approved in Japan in 2023, but only for retinal dystrophy with biallelic pathogenic variants in the RPE65 gene 9)18)19). Gene therapy for RP caused by other gene mutations, including RPGR mutations, is currently in clinical trials 7) and is not approved as a general treatment in Japan.
QHow can I receive investigational treatments?
A
Participation in clinical trials is limited to formal studies approved by the ethics committee of the medical institution. In addition to consulting with your physician, you can search for trial information on the Japan Registry of Clinical Trials (jRCT) operated by the National Cancer Center or on ClinicalTrials.gov in the United States.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.