จอประสาทตาเสื่อมชนิดสีสะสม (RP ) เป็นโรคจอประสาทตา เสื่อมทางพันธุกรรมที่พบใน 1 ใน 4,000–8,000 คน เป็นสาเหตุอันดับสอง (13.0%) ของความบกพร่องทางการมองเห็น ในผู้ที่ได้รับบัตรผู้พิการทางร่างกายรายใหม่อายุ ≥18 ปี และเป็นสาเหตุอันดับหนึ่งของภาวะตาบอดแต่กำเนิด 9) .
อาการแรกมักเป็นตาบอดกลางคืน (มองเห็นลำบากในที่มืด) ตามด้วยการแคบลงของลานสายตาอย่างค่อยเป็นค่อยไป โรคเริ่มมีอาการในช่วงอายุ 10–20 ปี และดำเนินไปจนถึงตาบอดใช้เวลาหลายสิบปี
มียีนเกี่ยวข้องมากกว่า 100 ยีน โดยมีรูปแบบการถ่ายทอดทางพันธุกรรมแบบออโตโซมอลโดมิแนนต์ ออโตโซมอลรีเซสซีฟ และแบบเชื่อมโยงกับโครโมโซม X กรณีประปรายพบมากที่สุด (48–63%) 9) .
ในชนิดรีเซสซีฟในคนญี่ปุ่น การกลายพันธุ์ของยีน EYS พบบ่อยที่สุด (30–50% ของกรณีที่ระบุยีนก่อโรคได้) 12) 13) .
ปัจจุบันยังไม่มีการรักษาที่หายขาด การดูแลมุ่งเน้นที่การปกป้องเซลล์รับแสง (วิตามินเอ ยูโนพรอสโตน นิลวาดิพีน ฯลฯ) การรักษาภาวะแทรกซ้อน (การรักษาภาวะบวมน้ำจอตาแบบซีสตอยด์ การผ่าตัดต้อกระจก ) และการดูแลผู้มีสายตาเลือนราง
ยาบำบัดด้วยยีน voretigene neparvovec (Luxturna) ได้รับการอนุมัติในญี่ปุ่นในปี 2023 9) จำกัดเฉพาะผู้ป่วยที่มีความแปรผันทางพยาธิวิทยาแบบสองอัลลีลในยีน RPE 65.
ได้รับการกำหนดให้เป็นโรคหายากที่กำหนด (ตั้งแต่วันที่ 1 มกราคม 2015) และมีสิทธิ์ได้รับเงินอุดหนุนค่ารักษาพยาบาล
จอประสาทตาเสื่อมชนิดสีสะสม (RP ) เป็นคำรวมสำหรับกลุ่มโรคทางพันธุกรรมที่มีลักษณะการเสื่อมแบบลุกลามเป็นวงกว้างของเซลล์รับแสง (เซลล์รูปแท่ง และเซลล์รูปกรวย ) และเยื่อบุผิวสีจอตา (RPE ) การเสื่อมของเซลล์รูปแท่ง เกิดขึ้นก่อนการเสื่อมของเซลล์รูปกรวย และชนิดนี้เรียกว่าโรคจอประสาทตา เสื่อมชนิดแท่ง-กรวย ซึ่งเข้าใจในความหมายเดียวกันกับ RP ไม่ใช่โรคเดียวแต่เป็นกลุ่มโรคที่เกี่ยวข้องกับยีนมากกว่า 100 ยีน
ความชุกคือ 1 ใน 4,000–8,000 คน โดยจำนวนผู้ป่วยทั้งหมดในญี่ปุ่นอย่างน้อย 30,000 คน (ผู้รับสิทธิโรคหายากที่กำหนด 20,687 คนในปี 2023) 9) ในส่วนที่เกี่ยวข้องกับความบกพร่องทางการมองเห็น RP เป็นสาเหตุอันดับสอง (13.0% รองจากต้อหิน 40.7% ในปี 2019) ของความบกพร่องทางการมองเห็น ในผู้ที่ได้รับบัตรผู้พิการทางร่างกายรายใหม่อายุ ≥18 ปี และเป็นสาเหตุอันดับหนึ่งของภาวะตาบอดแต่กำเนิด 9) ได้รับการกำหนดให้เป็นโรคหายากที่กำหนดภายใต้พระราชบัญญัติโรคหายากของญี่ปุ่น (ตั้งแต่วันที่ 1 มกราคม 2015) 9) และมีสิทธิ์ได้รับเงินอุดหนุนค่ารักษาพยาบาล
รูปแบบการถ่ายทอดทางพันธุกรรมแบ่งออกเป็นประเภทต่างๆ ดังนี้9) .
รูปแบบการถ่ายทอด ความถี่ ลักษณะ กรณีประปราย 48-63% พบบ่อยที่สุด; รวมถึง AR จำนวนมาก ถ่ายทอดแบบด้อยบนออโตโซม (AR) 20-35% การกลายพันธุ์ของ EYS พบบ่อยที่สุด ถ่ายทอดแบบเด่นบนออโตโซม (AD) 10-23% เริ่มมีอาการช้า พยากรณ์โรคค่อนข้างดี ถ่ายทอดแบบเชื่อมโยงกับโครโมโซม X (XL) 1.5-5% เพศชายรุนแรงกว่า; ดำเนินโรคเร็ว
เชื่อกันว่ากรณีที่เกิดขึ้นประปรายนั้นรวมถึงชนิด AR (ถ่ายทอดทาง autosomal recessive) จำนวนมาก
RP ยังมีชนิดที่เป็นกลุ่มอาการร่วมกับโรคทางระบบอื่นๆ และจัดอยู่ภายใต้แนวคิดที่กว้างขึ้นคือ โรคซิเลีย (ciliopathy) ดังนี้ 9) 2)
โรคซิเลีย (ciliopathy):
กลุ่มอาการอัชเชอร์ (type 1/2/3):RP + การสูญเสียการได้ยิน; โรคหายากที่กำหนด (AR). ใน type 1 จะมีการสูญเสียการได้ยินรุนแรงและความผิดปกติของระบบการทรงตัวตั้งแต่วัยเด็กกลุ่มอาการบาร์เด็ต-บีเดิล: โรคอ้วน, พัฒนาการทางจิตช้า, นิ้วเกิน, อวัยวะเพศเจริญไม่เต็มที่ (AR)กลุ่มอาการซีเนียร์-โลเกน: RP + โรคไตอักเสบในเด็กและวัยรุ่น (AR)กลุ่มอาการอัลสตรอม: RP + โรคอ้วน, การสูญเสียการได้ยิน, เบาหวาน (AR)กลุ่มอาการจูเบิร์ต: RP + สมองน้อยส่วน vermis เจริญไม่เต็มที่ (AR)
ความผิดปกติแต่กำเนิดของเมแทบอลิซึม:
โรคเมือกโพลีแซ็กคาไรด์สะสม (Hurler, Hunter): ร่วมกับความขุ่นของจอตาโรคเรฟซัม (ชนิดผู้ใหญ่และทารก): โรคเพอรอกซิโซม ; การเคลื่อนไหวไม่ประสานของสมองน้อย, โรคเส้นประสาทหลายเส้น (AR)กลุ่มอาการบาสเซน-คอร์นซ์ไวก์: ความผิดปกติของเมแทบอลิซึมไขมัน
โรคไมโทคอนเดรีย:
กลุ่มอาการเคิร์นส์-เซย์รี: กล้ามเนื้อตาภายนอกอ่อนแรงแบบลุกลามทั้งสองข้าง, หนังตาตก , การนำไฟฟ้าหัวใจผิดปกติ
โรคกล้ามเนื้อเสื่อม :
โรคกล้ามเนื้อเสื่อมชนิดไมโอโทนิก : อาจเกิดร่วมกับ RP
นอกจากนี้ การแยกความแตกต่างจากกลุ่มอาการต่างๆ เช่น PHARC (โรคปลายประสาทอักเสบหลายเส้น, หูหนวก, การทรงตัวผิดปกติ, RP , ต้อกระจก ), PCARP และกลุ่มอาการโอลิเวอร์-แมคฟาร์เลนก็มีความสำคัญเช่นกัน 3)
Q
โรคจอประสาทตาเสื่อมชนิดรงควัตถุเป็นโรคทางพันธุกรรมหรือไม่?
A
RP เป็นโรคทางพันธุกรรม แต่ไม่ได้ถ่ายทอดไปยังทุกคนเสมอไป ความเสี่ยงในการถ่ายทอดไปยังบุตรแตกต่างกันไปตามรูปแบบการถ่ายทอดทางพันธุกรรม ในชนิด AD มีโอกาส 50% ที่บุตรจะได้รับโรค ในขณะที่ชนิด AR หรือ XL ความเสี่ยงจะแตกต่างกันไปตามรูปแบบ การเกิดแบบประปราย (48-63% ของทั้งหมด) มักมีความเสี่ยงในการถ่ายทอดไปยังรุ่นต่อไปค่อนข้างต่ำ 9) แนะนำให้ใช้การให้คำปรึกษาทางพันธุกรรม
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MF
RP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PM
CI D: PMC2742641. License: CC BY.
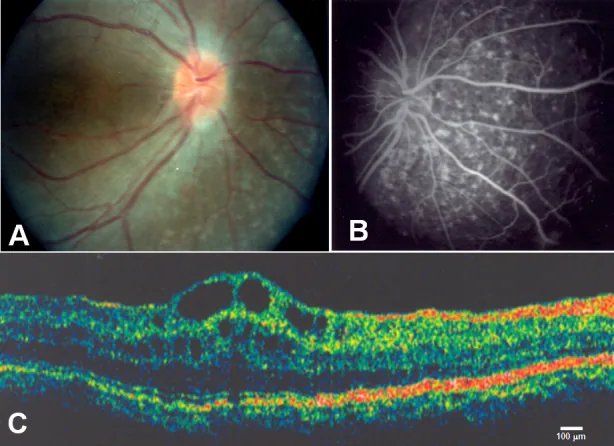
A แสดงดรูเซน ของหัวประสาทตาและการฝ่อของเยื่อบุผิวรงควัตถุจอประสาทตา อย่างกว้างขวาง, B แสดงการเรืองแสงของคอรอยด์ ที่สอดคล้องกับการฝ่อของเยื่อบุผิวรงควัตถุจอประสาทตา , C แสดงอาการบวมน้ำที่จุดรับภาพชนิดถุงน้ำและการแยกชั้นในของจอประสาทตา ที่รอยบุ๋มจอประสาทตา สอดคล้องกับอาการบวมน้ำที่จุดรับภาพชนิดถุงน้ำที่กล่าวถึงในหัวข้อ “2. อาการหลักและผลการตรวจทางคลินิก”
อาการของ RP เปลี่ยนแปลงไปตามระยะของโรค เนื่องจากเซลล์รูปแท่ง เสื่อมก่อน อาการตามัวในที่มืดจึงปรากฏเป็นอาการแรกเริ่ม
ตามัวในที่มืด (night blindness) : การมองเห็น ลดลงหรือมองไม่เห็นในที่มืด ปรากฏเร็วมากเนื่องจากเซลล์รูปแท่ง เสื่อมก่อน 9) ; รับรู้ได้ในช่วงอายุ 10-20 ปี ว่ามองเห็นลำบากในที่แสงสลัว ในระยะแรก การมองเห็น ในเวลากลางวันมักปกติการแคบลงของลานสายตา : ลานสายตาแคบลงทีละน้อยจากรอบนอกเข้าสู่ส่วนกลาง พัฒนาจากจุดบอดรูปวงแหวนไปสู่การแคบลงของลานสายตาแบบศูนย์กลาง (การมองเห็น แบบอุโมงค์) 9) การมองเห็น ลดลงเซลล์รูปกรวย ดำเนินต่อไปหลังจากเซลล์รูปแท่ง การมองเห็น ส่วนกลางก็ลดลงเช่นกัน หากมี CME ร่วมด้วย อาจเกิดการมองเห็น ลดลงระดับปานกลางในระยะค่อนข้างต้น ในบางกรณี การมองเห็น ส่วนกลางอาจคงอยู่จนถึงระยะท้ายกลัวแสง (ตาบอดกลางวัน) : ความไวต่อแสง มากเกินไป เป็นอาการของความผิดปกติของเซลล์รูปกรวย เพิ่มขึ้นเมื่อการเสื่อมของเซลล์รูปกรวย ดำเนินไป สิ่งสำคัญคือต้องแยกความแตกต่างจากการกระจายแสงที่เกิดจากต้อกระจก ภาพแสงวาบ (photopsia) : อาจเกิดขึ้นเนื่องจากการเสื่อมและการสูญเสียเซลล์รับแสง ภาพหลอนทางตา (Charles Bonnet syndrome) : ปรากฏการณ์ที่ผู้ป่วยที่มีการมองเห็น ลดลงเห็นทิวทัศน์หรือบุคคลที่ไม่มีอยู่จริง ไม่ใช่ประสบการณ์ทางพยาธิวิทยา แต่เป็นปรากฏการณ์จากการทำงานมากเกินไปของคอร์เทกซ์การเห็น9)
ด้านล่างเป็นแนวทางการดำเนินของอาการตามระยะของโรค
ระยะ อาการหลัก ช่วงอายุโดยประมาณ ระยะแรก ตาบอดกลางคืน 10–20 ปี ระยะกลาง การแคบลงของลานสายตา (scotoma วงแหวน → เข้าสู่ศูนย์กลาง) 30–40 ปี ระยะปลาย การมองเห็น ลดลง, ความผิดปกติของการมองเห็นสี , กลัวแสง50 ปีขึ้นไป
กลุ่มอาการคลาสสิกสามประการของ RP เป็นที่รู้จักดังนี้
เม็ดสีคล้ายกระดูกชิ้นเล็ก : เม็ดสีลักษณะเฉพาะที่ปรากฏจากบริเวณกึ่งกลางรอบนอกไปจนถึงรอบนอก (รูปแบบ bone spicule)หลอดเลือดแดงจอประสาทตา ตีบแคบ : เกิดขึ้นทุติยภูมิจากการเสื่อมของเซลล์รับแสง จานประสาทตา ซีดคล้ายขี้ผึ้งเส้นประสาทตา
การจำแนกชนิดของโรค แบ่งเป็นชนิดปกติและชนิดผิดปกติ 9) .
RP ชนิดปกติ (rod-cone dystrophy )เซลล์รูปแท่ง ถูกทำลายก่อน ตามด้วยเซลล์รูปกรวย ในภายหลัง
Rod dystrophy (ชนิดย่อย) : เซลล์รูปกรวย ไม่ถูกทำลายจนถึงระยะท้าย; การมองเห็น ส่วนกลางยังคงดีแม้ลานสายตาตีบรุนแรง
RP ชนิดผิดปกติ9) :
RP ไร้เม็ดสี: ไม่พบเม็ดสีRP ข้างเดียว: พบเพียงข้างเดียว หรือความแตกต่างระหว่างสองตามากRP เฉพาะส่วน: จำกัดอยู่ที่ 1-2 จตุภาคของจอประสาทตา ; ดำเนินโรคช้า พยากรณ์โรคดีRP ชนิดกลางและกึ่งกลาง: รอยโรคจอประสาทตา และความผิดปกติของลานสายตาเริ่มจากศูนย์กลางจอประสาทตา อักเสบชนิดจุดขาว: รอยโรคจุดสีขาวถึงเหลืองบนจอประสาทตา
ในเด็ก มักไม่พบลักษณะที่ครบถ้วน และ ERG เป็นกุญแจสำคัญในการวินิจฉัย
ภาวะแทรกซ้อนที่สำคัญมีดังนี้9)
ต้อกระจก ชนิดใต้แคปซูลด้านหลัง (PSC)การมองเห็น ลดลงในที่สว่าง ความกว้างของโซนรูปไข่ (EZ ) ≥600 ไมครอน สามารถทำนายการมองเห็น หลังผ่าตัดต้อกระจก ที่ดีได้ (AUC 0.97)5) จอประสาทตา บวมน้ำชนิดซีสตอยด์ (CME )การมองเห็น ส่วนกลางลดลง9) ต้อหินมุมปิด 9) เยื่อเหนือจอตา (Epiretinal membrane) : พบร่วม 15.6-27.3%9) รูจอตาและรอยแยกที่โฟเวีย : พบได้ค่อนข้างน้อย แต่อาจเป็นข้อบ่งชี้ในการผ่าตัดน้ำวุ้นตา 9)
Q
การมองเห็นดีขึ้นหลังผ่าตัดต้อกระจกหรือไม่?
A
ในการผ่าตัดต้อกระจก ชนิดใต้แคปซูลด้านหลังที่เกิดร่วมกับ RP หากความกว้างของโซนรูปไข่ (EZ ) จากการตรวจ OCT ก่อนผ่าตัด ≥600 ไมครอน สามารถคาดหวังการมองเห็น หลังผ่าตัดที่ดีได้ (AUC 0.97)5) ความกว้างของ EZ เป็นตัวบ่งชี้ทางชีวภาพ ที่มีประโยชน์ในการทำนายการทำงานของการมองเห็น ก่อนผ่าตัด อย่างไรก็ตาม เส้นใยซินน์มักจะอ่อนแอ จึงต้องระวังการหดรัดตัวของแคปซูลด้านหน้าและเลนส์แก้วตาเทียม เคลื่อน ในการป้องกัน CME หลังผ่าตัด (10-14%) แนะนำให้ใช้ยาหยอดตาสเตียรอยด์ และ NSAID เป็นระยะเวลานานกว่าปกติ9)
RP เป็นกลุ่มโรคที่มีความหลากหลายทางพันธุกรรมสูง เกิดจากการกลายพันธุ์ของยีนมากกว่า 100 ยีน9) ด้านล่างนี้คือยีนก่อโรคหลักในคนญี่ปุ่นจำแนกตามรูปแบบการถ่ายทอดทางพันธุกรรม
การเปรียบเทียบยีนก่อโรคหลักแสดงไว้ด้านล่าง
ยีน รูปแบบการถ่ายทอด ความถี่/ลักษณะเฉพาะในคนญี่ปุ่น EYS AR 30-50% ของกรณีที่ระบุยีนก่อโรคได้ (พบมากที่สุดในชนิด AR)12) 13) USH2A AR พบบ่อยเป็นอันดับสองในชนิด AR (4-9%); ยีนหลักของกลุ่มอาการอัชเชอร์ 12) RHO AD พบบ่อยที่สุดในชนิด AD 6) RP GRเชื่อมโยงกับโครโมโซม X ประมาณ 70-75% ในชนิด XL 6) REEP6 AR หนึ่งในยีนก่อโรคชนิด AR 4)
รายละเอียดเพิ่มเติมเกี่ยวกับลักษณะของยีนแต่ละชนิดมีดังนี้
EYS (Eyes Shut Homolog) : ยีนก่อโรคที่พบบ่อยที่สุดในชนิด AR RP ในคนญี่ปุ่น (30-50% ของผู้ป่วยที่ระบุยีนได้) 12) 13) ความถี่นี้ไม่สูงเท่าในประเทศตะวันตก สะท้อนถึงพื้นฐานทางพันธุกรรมที่เป็นเอกลักษณ์ของคนญี่ปุ่นUSH2A : ยีนหลักของกลุ่มอาการอัชเชอร์ (RP + สูญเสียการได้ยิน) และเป็นยีนที่พบบ่อยเป็นอันดับสองในชนิด AR RP ในคนญี่ปุ่นรองจาก EYS (4-9%) 12) RHO (Rhodopsin) : ยีนที่พบบ่อยที่สุดที่ทำให้เกิด RP ชนิด AD 6) กำหนดรหัสโปรตีนรับแสงในเซลล์รูปแท่ง RP GR (Retinitis Pigmentosa GTPase Regulator)RP ชนิด XL 6) มีรายงานผู้ป่วยชายที่มีการกลายพันธุ์ของ RP GR และมีภาวะผิดปกติของซิเลียปฐมภูมิ (PCD) ร่วมด้วย 1) REEP6 (Receptor Expression-Enhancing Protein 6) : หนึ่งในยีนที่ทำให้เกิด RP ชนิด AR 4)
อัตราการตรวจพบยีนก่อโรคในการตรวจทางพันธุกรรมแตกต่างกันไปตามรูปแบบการถ่ายทอดทางพันธุกรรม สำหรับชนิด AD 35-60%, ชนิด AR/ผู้ป่วยประปราย 30-50%, และชนิด XL 16-36% 6)
นอกจากนี้ ใน RP ที่มีกลุ่มอาการ2) การแยกโรคจากกลุ่มอาการต่างๆ เช่น ภาวะเสียการทรงตัวของฟรีดริช (PHARC), PCARP , กลุ่มอาการ Oliver-McFarlane เป็นสิ่งสำคัญ 3)
Q
ควรตรวจพันธุกรรมหรือไม่?
A
การวินิจฉัยทางพันธุกรรมมีความสำคัญต่อการวินิจฉัยที่แน่นอน การให้คำปรึกษาทางพันธุกรรม และการพิจารณาความเหมาะสมในการรักษาด้วยยีน ระบบชุดตรวจ PrismGuide IRD (การวิเคราะห์ลำดับเอ็กซอนของยีนก่อโรคจอประสาทตา ทางพันธุกรรม 82 ยีนอย่างครอบคลุม) ได้รับการอนุมัติให้ครอบคลุมโดยประกันในปี 2023 แต่ ณ เดือนมิถุนายน 2025 มีไว้สำหรับผู้ป่วยอายุน้อยที่สงสัยว่าเป็น IRD ที่เกี่ยวข้องกับ RPE 65 เท่านั้น 9) แนะนำให้ทำร่วมกับการให้คำปรึกษาทางพันธุกรรม สามารถรับคำปรึกษาทางพันธุกรรมได้โดยไม่ต้องตรวจวินิจฉัยทางพันธุกรรม
การวินิจฉัย RP ทำได้โดยการรวมผลการตรวจทางคลินิก การตรวจทางไฟฟ้าสรีรวิทยา การตรวจภาพ และการตรวจทางพันธุกรรม
เกณฑ์การวินิจฉัย (เกณฑ์การรับรอง) 9) ประกอบด้วยองค์ประกอบต่อไปนี้:
ก. อาการ (อย่างน้อย 1 ข้อ) :
อาการที่ผู้ป่วยรู้สึกได้เองและดำเนินไป
อย่างน้อย 1 ข้อจาก: ตาบอดกลางคืน , การมองเห็น แคบลง, สายตาเลือนลาง, กลัวแสง
ข. ผลการตรวจ (อย่างน้อย 2 ข้อ) :
(1) ผลการตรวจอวัยวะภายในตา: หลอดเลือดจอประสาทตา ตีบ, สีจอประสาทตา หยาบ, เม็ดสีรูปกระดูก, จุดขาวหลายจุด, เส้นประสาทตา ฝ่อ, จุดรับภาพเสื่อม
(2) ความผิดปกติของ ERG
(3) ความผิดปกติของผล FAF
(4) ความผิดปกติของ EZ (ellipsoid zone) ใน OCT
ค. การตรวจทางพันธุกรรม (เสริม)
ง. การแยกโรคอื่นออก
การจำแนกระดับความรุนแรง 9) 10) :
ระดับ I : สายตาที่แก้ไขแล้ว ≥ 0.7, ไม่มีการแคบของลานสายตาระดับ II : สายตาที่แก้ไขแล้ว ≥ 0.7, มีการแคบของลานสายตา (เข้าข่ายโรคหายากที่กำหนด)ระดับ III : สายตาที่แก้ไขแล้ว < 0.7 และ ≥ 0.2 (เข้าข่ายโรคหายากที่กำหนด)ระดับ IV : สายตาที่แก้ไขแล้ว < 0.2 (เข้าข่ายโรคหายากที่กำหนด)
วิธีการตรวจหลักแสดงไว้ด้านล่าง
การตรวจ บทบาทหลัก ข้อสังเกตพิเศษ การตรวจคลื่นไฟฟ้าจอตา (ERG ) การวินิจฉัยที่แน่นอน การตอบสนองของเซลล์รูปแท่ง ลดลงก่อนการตอบสนองของเซลล์รูปกรวย 6) 9) OCT การประเมินสภาพและการพยากรณ์โรค ความกว้าง EZ : ตัวบ่งชี้ทางชีวภาพ สำหรับการพยากรณ์5) ; การบางลงของชั้นเม็ดเล็กด้านนอก FAF การประเมินกิจกรรมของโรค วงแหวนเรืองแสงผิดปกติ (AF ring) เป็นตัวบ่งชี้ระยะโรค6) การตรวจลานสายตา การประเมินการดำเนินโรค เครื่องวัดลานสายตา Goldmann; โปรแกรม HFA 10-2 ก็มีประโยชน์9) 10) การตรวจการมองเห็น สี การประเมินการทำงานของเซลล์รูปกรวย ความผิดปกติแบบน้ำเงิน-เหลืองที่เกิดขึ้นภายหลังพบบ่อย; การทดสอบ Panel D-15 และ 100 Hue9) การทดสอบการปรับตัวในที่มืด การประเมินการทำงานของเซลล์รูปแท่ง ไม่พบจุดหักเหของ Kohlrausch10) การตรวจยีน NGS การวินิจฉัยทางพันธุกรรม ชุดตรวจ PrismGuide IRD (82 ยีน) 9)
รายละเอียดของการตรวจแต่ละรายการแสดงไว้ด้านล่าง
การตรวจคลื่นไฟฟ้าจอตา (ERG ) : จำเป็นสำหรับการวินิจฉัยที่แน่นอน 6) 9) การตอบสนองของเซลล์รูปแท่ง (ERG ในที่มืด) ลดลงตั้งแต่แรกเริ่ม และเมื่อโรคดำเนินไป การตอบสนองของเซลล์รูปกรวย (ERG ในที่สว่าง) ก็ลดลงเช่นกัน ERG แบบเต็มลานตาเป็นมาตรฐานพื้นฐาน มักจะไม่สามารถบันทึกสัญญาณได้แล้วเมื่อมาตรวจครั้งแรกOCT (เครื่องตรวจชั้นจอตาด้วยแสง)EZ (โซนรูปไข่) ความกว้างของ EZ มีประโยชน์เป็นตัวบ่งชี้ทางชีวภาพ เชิงปริมาณสำหรับการทำงานของการมองเห็น และการพยากรณ์โรค และยังใช้ในการพิจารณาความเหมาะสมของการผ่าตัดต้อกระจก 5) พบการบางลงของชั้นนิวเคลียสชั้นนอกและการหายไปของ EZ ตั้งแต่ระยะแรกการตรวจการเรืองแสงเองของจอตา (FAF ) : พบวงแหวนเรืองแสงเองผิดปกติ (วงแหวน AF) รอบจอประสาทตา ซึ่งเป็นตัวบ่งชี้การดำเนินโรคและจอตาที่ยังมีหน้าที่เหลืออยู่ 6) การตรวจลานสายตา การตรวจลานสายตา แบบพลวัตด้วยเครื่องวัดลานสายตา Goldmann เป็นมาตรฐาน เมื่อโรคดำเนินไป จะเป็นไปตามแบบ scotoma วงแหวน → การแคบเข้าหาศูนย์กลางของลานสายตา 10) โปรแกรม HFA 10-2 มีประโยชน์ในการประเมินการทำงานของเซลล์รูปกรวย ส่วนกลางที่เหลืออยู่ 9) การตรวจการมองเห็น สี : มักพบความผิดปกติแบบสีน้ำเงิน-เหลืองที่เกิดขึ้นภายหลัง ประเมินด้วยการทดสอบ Panel D-15 และการทดสอบ 100 Hue 9) การตรวจการปรับตัวในที่มืด : ไม่พบจุดหักเหของ Kohlrausch (จุดเปลี่ยนระหว่างเซลล์รูปแท่ง และเซลล์รูปกรวย ) 10) เครื่องหาลำดับเบสรุ่นใหม่ (NGS) : ระบบชุดตรวจ PrismGuide IRD สามารถวิเคราะห์ลำดับเอ็กซอนของยีนก่อโรค 82 ยีนได้อย่างครอบคลุม 9) นอกจากนี้ยังจำเป็นสำหรับการพิจารณาความเหมาะสมของการรักษาด้วยยีน
โรคที่ต้องวินิจฉัยแยกโรคหลักตาม GL2026 แสดงไว้ด้านล่าง 9)
โรคทางพันธุกรรม : จอประสาทตา เสื่อมชนิดเซลล์รูปกรวย , จอประสาทตา เสื่อมชนิดเซลล์รูปกรวย -แท่ง, โรคสตาร์การ์ดต์ (ยีน ABCA4; จอประสาทตา เสื่อมเฉพาะที่), โรคโอกุจิ (จอตาดุจแผ่นทองคำ; ปรากฏการณ์มิซูโอะ-นากามูระ), ตามัวในที่มืดแต่กำเนิดชนิดคงที่ (จอตาปกติถึงสายตาสั้น ; คลื่น b ลบ), จอตามีจุดขาวกระจาย (fundus albipunctatus; ยีน RDH5), จอตาฉีกขาดแต่กำเนิด (ชนิด XL; เพศชาย), คอรอยเดอเรเมีย (คอรอยด์ ฝ่อกระจาย; ยีน CHM), จอตาและคอรอยด์ ฝ่อแบบวงแหวน (ornithine ในเลือดสูง; OAT บกพร่อง), จอตาเสื่อมแบบผลึก
โรคที่เกิดขึ้นภายหลัง : จอประสาทตา อักเสบจากภูมิต้านตนเอง (AIR), จอประสาทตาเสื่อมที่สัมพันธ์กับมะเร็ง (CAR), จอประสาทตาเสื่อมที่สัมพันธ์กับมะเร็ง ผิวหนังชนิดเมลาโนมา (MAR) (เริ่มต้นค่อนข้างเฉียบพลัน; ค้นหาแอนติบอดีต่อจอตา), จอประสาทตาเสื่อมจากยา (คลอโรควิน, เมลาโทนิน ฯลฯ), จอประสาทตา เสื่อมจากการบาดเจ็บ, การติดเชื้อ (หัดเยอรมัน, ซิฟิลิส, ทอกโซพลาสมา), AZOOR (ลานสายตาบอดเฉียบพลัน; จอตาปกติในระยะแรก)
ปัจจุบันยังไม่มีการรักษาที่หายขาดสำหรับ RP 6) 9) การรักษามุ่งเน้นที่การรักษาการทำงานของการมองเห็น การจัดการภาวะแทรกซ้อน และการสนับสนุนการใช้ชีวิตในสังคม
การปกป้องเซลล์รับแสง
วิตามินเอ (15,000 IU/วัน) : มีรายงานว่าการรับประทานชะลอการเสื่อมของ ERG ได้ไม่กี่เปอร์เซ็นต์ 14) ไม่ช่วยปรับปรุงการมองเห็น หรือลานสายตา จำเป็นต้องติดตามการทำงานของตับเมื่อใช้ระยะยาว ห้ามใช้ในระหว่างตั้งครรภ์เนื่องจากเสี่ยงต่อการทำให้ทารกพิการ ในผู้ที่มีการกลายพันธุ์ ABCA4 อาจทำให้โรคดำเนินเร็วขึ้น 14) ข้อควรระวัง: วิตามินอีอาจทำให้โรคดำเนินเร็วขึ้น จึงต้องระมัดระวัง 14)
ยาหยอดตา Unoprostone : แสดงแนวโน้มการปรับปรุงความไวที่ขึ้นกับขนาดยา แต่ไม่มีนัยสำคัญทางสถิติในตัวชี้วัดหลักของการทดลองระยะที่ 2 (ความไวของจอประสาทตา ส่วนกลาง 2 องศา) 16)
Nilvadipine (ยาปิดกั้นช่องแคลเซียม) : รายงานระยะยาวบ่งชี้ว่าชะลออัตราการดำเนินของความบกพร่องของลานสายตา 15) รายงานเหล่านี้มาจากศูนย์เดียวและมีจำนวนผู้ป่วยน้อย ยังไม่มีการทดลองแบบหลายศูนย์
N-acetylcysteine (NAC) : ยับยั้งความเครียดออกซิเดชัน การทดลองระยะที่ 1 รายงานว่าช่วยปรับปรุงการมองเห็น 17) และกำลังมีการทดลองระยะที่ 3 ในปี 2025
DHA และลูทีน : ปกป้องเซลล์รับแสง บริเวณจอประสาทตา ส่วนกลางจากความเครียดออกซิเดชัน การเพิ่ม DHA ลงในวิตามินเอไม่พบประโยชน์เพิ่มเติม
Helenien (Adaptinol) : ได้รับการอนุมัติให้ใช้ปรับปรุงลานสายตาและการปรับตัวในที่มืดชั่วคราวใน RP ยังไม่มีการประเมินประสิทธิผลตามมาตรฐานการแพทย์สมัยใหม่
แว่นตาป้องกันแสง : ลดความเครียดออกซิเดชันจากรังสียูวีและแสงจ้า แนะนำให้ใช้ทุกวัน
การรักษาภาวะแทรกซ้อน
การรักษาภาวะจอประสาทตา บวมน้ำชนิดซีสต์ (CME ) :
ทางเลือกแรกคือยับยั้งเอนไซม์คาร์บอนิกแอนไฮเดรส (CAI)
ใช้ dorzolamide (Trusopt) ยาหยอดตาหรือ acetazolamide (Diamox) รับประทาน การปรับปรุงความหนาจอประสาทตา ส่วนกลาง (CMT) ทำได้ประมาณ 40% เกิดการกลับเป็นซ้ำประมาณ 30% 9)
หากดื้อต่อ CAI ให้พิจารณาสเตียรอยด์
ใช้ triamcinolone acetonide (Macuoid) ฉีดเข้าแก้วตา หรือ dexamethasone ฝังในแก้วตา (Ozurdex )
ไม่แนะนำให้ใช้ยาต้าน VEGF ใน RP -CME เนื่องจากการผลิต VEGF ลดลง 9)
โปรดทราบว่าไม่มีวิธีการใดที่ได้รับการอนุมัติจากประกันสำหรับ RP -CME และใช้เป็นการใช้นอกเหนือข้อบ่งชี้
การผ่าตัดต้อกระจก : ดำเนินการในผู้ป่วยที่มีต้อกระจก ชนิดใต้แคปซูลด้านหลัง ความกว้างของ EZ ≥600 μm จากการตรวจ OCT ก่อนผ่าตัดเป็นปัจจัยพยากรณ์การมองเห็น หลังผ่าตัดที่ดี 5) ในกรณีที่เอ็นซินน์อ่อนแอ ให้พิจารณาใช้วงแหวนขยายแคปซูลเลนส์ เพื่อป้องกัน CME หลังผ่าตัด (10-14%) ให้ใช้ยาหยอดตาสเตียรอยด์ และ NSAID เป็นระยะเวลานานกว่าปกติ 9)
ต้อหินมุมปิด ต้อหินมุมปิดปฐมภูมิ สูงในผู้ป่วย RP ช่องหน้าม่านตา จะตื้นขึ้นเรื่อย ๆ ทำการตัดม่านตาด้วยเลเซอร์ เพื่อป้องกันหรือผ่าตัดต้อกระจก 9)
เยื่อเหนือจอตา (GL2026 CQ4) : การตัดแก้วตา การมองเห็น ดีขึ้นได้ในกรณีที่เส้น EZ ต่อเนื่อง ในกรณีที่เส้น EZ ไม่ต่อเนื่อง การฟื้นตัวมีจำกัด มีรายงานการฝ่อของจอประสาทตา อย่างรุนแรงในระยะยาวหลังผ่าตัด ดังนั้นควรประเมินในสถานพยาบาลเฉพาะทาง 9)
รูที่จอตา : การตัดแก้วตาเป็นการรักษาที่หายขาดเพียงวิธีเดียว การศึกษาเกี่ยวกับผลลัพธ์หลังผ่าตัดมีจำกัด 9)
การสนับสนุนและการฟื้นฟู
การดูแลผู้มีสายตาเลือนราง การมองเห็น เป็นสิ่งสำคัญ แนะนำให้ใช้ Smart Site (แนะนำสถานที่ปรึกษาสายตาเลือนรางในแต่ละภูมิภาค)
การให้คำปรึกษาทางพันธุกรรม
ระบบโรคหายาก : สามารถใช้ระบบช่วยเหลือค่าใช้จ่ายทางการแพทย์สำหรับโรคหายากที่กำหนด 9) ควรพิจารณาขอบัตรประจำตัวคนพิการทางกายและการสนับสนุนการดูแลตนเองด้วย
Voretigene neparvovec (Luxturna) : ยารักษาด้วยยีนที่สามารถให้แก่ผู้ป่วยที่มีความแปรปรวนทางพยาธิแบบสองอัลลีลในยีน RPE 65 และมีเซลล์จอประสาทตา ที่มีชีวิตเพียงพอ ได้รับการอนุมัติในญี่ปุ่นในปี 2023 9) ในการทดลองระยะที่ 3 ของสหรัฐอเมริกา (การศึกษา 301) มีผู้ลงทะเบียน 31 ราย และการวิเคราะห์ mITT (กลุ่มแทรกแซง 20 ราย กลุ่มควบคุม 9 ราย) แสดงให้เห็นการปรับปรุงอย่างมีนัยสำคัญใน MLMT และ FST แสงสีขาวเมื่อเทียบกับกลุ่มควบคุม 18) ในการทดลองระยะที่ 3 ในประเทศ (การศึกษา A11301) พบว่าความไว FST เพิ่มขึ้นและลานสายตาขยายในผู้ป่วยญี่ปุ่น 4 ราย 19) ผลในการปรับปรุงการมองเห็น มีจำกัด และมีรายงานการฝ่อของคอรอยด์ และจอประสาทตา เป็นภาวะแทรกซ้อนระยะยาวในมากกว่า 20% 9)
ดำเนินการทุก 6 เดือนถึง 1 ปี: การมองเห็น , กล้องจุลทรรศน์ชนิดกรีด, จอตา, การวัดลานสายตา Humphrey (HFA 10-2), OCT 9)
การรับประทานวิตามินเอมากเกินไปมีความเสี่ยงต่อพิษต่อตับและทำให้ทารกพิการ อย่าเพิ่มขนาดยาเอง 14)
ในจีโนไทป์เช่นการกลายพันธุ์ ABCA4 วิตามินเออาจส่งเสริมการดำเนินโรค ดังนั้นจึงสำคัญที่จะปรึกษาผู้เชี่ยวชาญหลังการวินิจฉัยทางพันธุกรรม 14)
การฉีดสเตียรอยด์ เข้าแก้วตาเพื่อรักษาภาวะจอประสาทตา บวมน้ำชนิดถุงน้ำมีความเสี่ยงต่อความดันลูกตา สูงและการดำเนินของต้อกระจก
ข้อบ่งชี้ในการผ่าตัดต้อกระจก จะพิจารณาหลังจากการประเมิน ERG และความกว้างของ EZ อย่างรอบคอบ
ไม่มียาใดสำหรับรักษาภาวะจอประสาทตา บวมน้ำชนิดถุงน้ำที่ได้รับการอนุมัติจากประกันสุขภาพสำหรับ RP -CME ดังนั้นจึงเป็นการใช้นอกข้อบ่งชี้ 9)
Q
ยาชนิดใดที่ใช้รักษาภาวะจอประสาทตาบวมน้ำ?
A
การรักษาทางเลือกแรกสำหรับ RP -CME คือยับยั้งเอนไซม์คาร์บอนิกแอนไฮเดรส เช่น ยาหยอดตาดอร์โซลาไมด์ หรือยาเม็ดอะเซตาโซลาไมด์ รับประทาน 9) การปรับปรุง CMT ทำได้ประมาณ 40% ของกรณี แต่ประมาณ 30% เกิดการกลับเป็นซ้ำ หากไม่ดีขึ้นด้วย CAI อาจเลือกใช้การฉีดไตรแอมซิโนโลน อะซีโทไนด์เข้าแก้วตา หรือการฝังเดกซาเมทาโซนเข้าแก้วตา (Ozurdex ) ไม่แนะนำให้ใช้ยา anti-VEGF สำหรับ RP -CME ควรทราบว่าทั้งหมดนี้เป็นการใช้นอกข้อบ่งชี้ที่ได้รับการอนุมัติ
วิถีร่วมสุดท้ายของการตายของเซลล์รับแสง ใน RP คืออะพอพโทซิส แม้ว่าชนิดของการกลายพันธุ์ทางพันธุกรรมจะหลากหลาย แต่สุดท้ายแล้วจะมาบรรจบกันที่วิถีการตายของเซลล์ร่วมกัน
ใน RP เซลล์รูปแท่ง จะเสื่อมและหายไปก่อน ตามด้วยการเสื่อมทุติยภูมิของเซลล์รูปกรวย 7) เซลล์รูปกรวย ต้องพึ่งพาปัจจัยโภชนาการที่ผลิตโดยเซลล์รูปแท่ง (RdCVF) เพื่อความอยู่รอด ดังนั้นหลังจากเซลล์รูปแท่ง หายไป เซลล์รูปกรวย ก็จะสูญเสียการทำงานเช่นกัน 7) 11)
จอประสาทตา เป็นเนื้อเยื่อที่มีการเผาผลาญสูงที่สุดชนิดหนึ่ง โดยเปลี่ยนกลูโคส 80-90% เป็นแลคเตทผ่านไกลโคไลซิสแบบใช้ออกซิเจน (Warburg effect) เซลล์รูปกรวย มีความไวต่อความเครียดจากการเผาผลาญมากกว่าเซลล์รูปแท่ง และความไวต่อการเผาผลาญนี้ยังเป็นปัจจัยในการเสื่อมทุติยภูมิของเซลล์รูปกรวย 11)
การอักเสบยังได้รับการยอมรับว่าเป็นปัจจัยหลักในการดำเนินของ RP โดยการกระตุ้นไมโครเกลียและการแทรกซึมของแมคโครฟาจทำให้ความเสียหายของจอประสาทตา แย่ลง 11) ความเครียดออกซิเดชันยังทำหน้าที่เป็นตัวขับเคลื่อนทางชีวภาพของการเสื่อมทุติยภูมิของเซลล์รูปกรวย
กลไกการเสื่อมแตกต่างกันไปตามยีน
การกลายพันธุ์ RHO : โรดอปซินที่พับผิดรูปทำให้เกิดความเครียดของเอนโดพลาสมิกเรติคูลัม → การตอบสนองต่อโปรตีนที่พับผิดรูป → อะพอพโทซิส 11) การกลายพันธุ์ REEP6 : REEP6 เข้ารหัสโปรตีนที่เกี่ยวข้องกับการรักษาสัณฐานของ ER การกลายพันธุ์ที่ก่อโรคทำให้เกิดการสร้าง inclusion ใน ER ที่ส่วนนอกของเซลล์รูปแท่ง นำไปสู่การเสื่อมของเซลล์รับแสง 4) การกลายพันธุ์ RP GR : RP GR เกี่ยวข้องกับโครงสร้าง axoneme ของซิเลียปฐมภูมิ และการกลายพันธุ์ทำให้การขนส่งสารไปยังส่วนนอกของเซลล์รับแสง บกพร่อง1)
เนื้อหาต่อไปนี้อยู่ในขั้นตอนการวิจัยหรือการทดลองทางคลินิก และไม่ใช่การรักษามาตรฐานที่สามารถรับได้ในโรงพยาบาลทั่วไป เป็นข้อมูลอ้างอิงสำหรับผู้เชี่ยวชาญเกี่ยวกับการพัฒนาทางการแพทย์ในอนาคต
การบำบัดด้วยยีน เป็นแนวทางที่มีแนวโน้มมากที่สุดในการรักษาโรคจอประสาทตา ทางพันธุกรรม8)
Luxturna (voretigene neparvovec) : ยารักษาด้วยยีนที่สามารถให้แก่ผู้ป่วยที่มีความแปรผันทางพยาธิวิทยาแบบสองอัลลีลในยีน RPE 65 ในการทดลองระยะที่ 3 ของสหรัฐอเมริกา (การศึกษา 301) มีผู้ลงทะเบียน 31 ราย และการวิเคราะห์ mITT (กลุ่มแทรกแซง 20 ราย กลุ่มควบคุม 9 ราย) แสดงให้เห็นการปรับปรุงอย่างมีนัยสำคัญใน MLMT และ FST แสงสีขาวเมื่อเทียบกับกลุ่มควบคุม18) ในการทดลองระยะที่ 3 ในประเทศ (การศึกษา A11301) มีการยืนยันการเพิ่มความไวของ FST และการขยายลานสายตาในชาวญี่ปุ่น 4 ราย19) ได้รับการอนุมัติในญี่ปุ่นในปี 2023 และทำหน้าที่เป็นสะพานเชื่อมระหว่างการรักษามาตรฐานและการรักษาวิจัย
การบำบัดด้วยยีน RP GRการบำบัดด้วยยีน ที่ใช้ AAV เป็นพาหะสำหรับ XL-RP เนื่องจากการกลายพันธุ์ RP GR ได้ก้าวหน้าไปสู่การทดลองทางคลินิกระยะที่ 1/2/38)
CRISPR/Cas9 : การวิจัยกำลังดำเนินการเพื่อแก้ไขการกลายพันธุ์ที่ก่อโรคโดยตรงหรือยับยั้งการกลายพันธุ์แบบเด่นเชิงลบ8)
RdCVF เป็นโปรตีนที่หลั่งจากเซลล์รูปแท่ง และรักษาการอยู่รอดของเซลล์รูปกรวย 7) 11) การทดลองทางคลินิกของการบำบัดปกป้องเซลล์รูปกรวย โดยใช้ RdCVF กำลังดำเนินอยู่ และเป็นกลยุทธ์การรักษาอิสระเพื่อรักษาการทำงานของเซลล์รูปกรวย หลังการเสื่อมของเซลล์รูปแท่ง
NAC เป็นยาที่ยับยั้งความเครียดออกซิเดชัน และการทดลองระยะที่ 1 รายงานว่ามีการปรับปรุงการมองเห็น 17) ณ ปี 2025 การทดลองระยะที่ 3 กำลังดำเนินอยู่
การศึกษาในร่างกายเมื่อเร็วๆ นี้ (แบบจำลองหนู rd10) แสดงให้เห็นว่า dexamethasone ชนิดฉีดเข้าแก้วตา ช่วยปกป้องเซลล์รับแสง รูปกรวยและเยื่อบุผิวเม็ดสีจอประสาทตา 11) กลูโคคอร์ติคอยด์มีศักยภาพในการนำกลับมาใช้ใหม่สูงในฐานะการรักษาที่ไม่ขึ้นกับการกลายพันธุ์ อย่างไรก็ตาม หลักฐานในปัจจุบันจำกัดอยู่เพียงแบบจำลองสัตว์ และจำเป็นต้องมีการตรวจสอบเพิ่มเติมสำหรับการประยุกต์ใช้ทางคลินิกในมนุษย์
การปลูกถ่ายจอประสาทตา จากเซลล์ iPS : การวิจัยเกี่ยวกับการปลูกถ่ายแผ่นเซลล์รับแสง ที่สร้างจากเซลล์ iPS ของผู้ป่วยเองกำลังดำเนินอยู่จอประสาทตา เทียม (อุปกรณ์เทียมจอประสาทตา )RP ระยะสุดท้าย Argus II และอื่นๆ ได้ถูกนำมาใช้จริงในต่างประเทศ และการทดลองทางคลินิกของวิธีการกระตุ้นผ่านคอรอยด์ -จอประสาทตา กำลังดำเนินอยู่ในญี่ปุ่น
Q
การบำบัดด้วยยีนมีให้บริการในญี่ปุ่นด้วยหรือไม่?
A
Voretigene neparvovec (Luxturna) ได้รับการอนุมัติในญี่ปุ่นในปี 2023 แต่จำกัดเฉพาะจอประสาทตา เสื่อมที่มีความแปรปรวนทางพยาธิวิทยาแบบสองอัลลีลในยีน RPE 65 9) 18) 19) การบำบัดด้วยยีน สำหรับ RP ที่มีการกลายพันธุ์ของยีนอื่นๆ รวมถึงการกลายพันธุ์ RP GR ยังอยู่ในขั้นตอนการทดลองทางคลินิก 7) และยังไม่ได้รับการอนุมัติให้เป็นการรักษาทั่วไปในญี่ปุ่น
Q
จะได้รับการรักษาในขั้นตอนการวิจัยได้อย่างไร?
A
การเข้าร่วมการทดลองทางคลินิกจำกัดเฉพาะการทดลองอย่างเป็นทางการที่ได้รับอนุมัติจากคณะกรรมการจริยธรรมของสถาบันการแพทย์ นอกจากการปรึกษาแพทย์ผู้รักษาแล้ว ยังสามารถค้นหาข้อมูลการทดลองได้จากเว็บไซต์ข้อมูลการทดลองทางคลินิก (jRCT) ที่ดำเนินการโดยศูนย์มะเร็งแห่งชาติ หรือ clinicaltrials.gov ของสหรัฐอเมริกา
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RP GR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCI D:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP , and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCI D:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCI D: PMC11352491. doi:10.3390/biom14080903.
厚生労働科学研究費補助金難治性疾患政策研究事業網膜脈絡膜・視神経萎縮症に関する調査研究班. 網膜色素変性診療ガイドライン2026. 日眼会誌. 2026.
厚生労働科学研究費補助金難治性疾患政策研究事業網膜脈絡膜・視神経萎縮症に関する調査研究班. 網膜色素変性診療ガイドライン(旧版). 日眼会誌.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCI D:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCI D:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCI D:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI 132990. PMID:31805012; PMCI D:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE 65v2) in patients with RPE 65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCI D:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE 65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCI D:PMC12405627.