Retinitis pigmentosa (RP) ist ein Sammelbegriff für eine Gruppe erblicher Erkrankungen, die durch eine fortschreitende, ausgedehnte Degeneration der Photorezeptoren (Stäbchen und Zapfen) und des retinalen Pigmentepithels (RPE) gekennzeichnet sind. Die Degeneration der Stäbchen geht der der Zapfen voraus; dies wird als Stäbchen-Zapfen-Dystrophie bezeichnet, und RP wird als Synonym dafür betrachtet. Es handelt sich nicht um eine einzelne Erkrankung, sondern um eine Gruppe von Erkrankungen, an denen über 100 Gene beteiligt sind.
Die Prävalenz liegt bei 1 zu 4.000 bis 8.000, und die Gesamtzahl der Patienten in Japan beträgt mindestens 30.000 (20.687 Leistungsempfänger der seltenen Erkrankung im Jahr 2023) 9). Im Zusammenhang mit Sehbehinderung ist sie die zweithäufigste Ursache für Sehbehinderung (13,0 %) bei Neuerwerbern eines Schwerbehindertenausweises ab 18 Jahren (nach Glaukom mit 40,7 % im Jahr 2019) und die häufigste Ursache für angeborene Blindheit 9). In Japan ist sie als seltene Erkrankung nach dem Gesetz über seltene Erkrankungen anerkannt (seit 1. Januar 2015) 9) und berechtigt zur Kostenübernahme für medizinische Behandlungen.
RP tritt auch als syndromale RP mit anderen systemischen Erkrankungen auf, und Ziliopathie wird als übergeordnetes Konzept wie folgt klassifiziert 9)2).
Ziliopathie (Ciliopathy) :
Usher-Syndrom (Typ 1/2/3) : RP + Hörverlust; designierte seltene Krankheit (AR). Typ 1 geht bereits im Kindesalter mit hochgradigem Hörverlust und vestibulären Funktionsstörungen einher.
Darüber hinaus ist die Abgrenzung zu verschiedenen Syndromen wie PHARC (Polyneuropathie, Schwerhörigkeit, Ataxie, RP, Katarakt), PCARP und Oliver-McFarlane-Syndrom wichtig3).
QIst Retinitis pigmentosa erblich?
A
RP ist eine Erbkrankheit, wird aber nicht zwangsläufig an alle Kinder vererbt. Das Vererbungsrisiko für Kinder hängt vom Vererbungsmodus ab. Bei autosomal-dominantem (AD) Erbgang besteht ein 50%iges Risiko für die Kinder, während bei autosomal-rezessivem (AR) oder X-chromosomalem (XL) Erbgang das Risiko je nach Modus variiert. Bei sporadischen Fällen (48–63 % aller Fälle) ist das Risiko für die nächste Generation oft relativ gering9). Die Inanspruchnahme einer genetischen Beratung wird empfohlen.
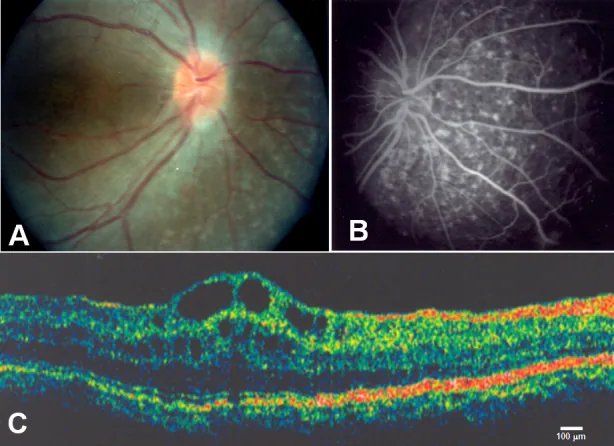
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A: Optikusdrusen und ausgedehnte Atrophie des retinalen Pigmentepithels; B: chorioidale Hyperfluoreszenz entsprechend der Atrophie des retinalen Pigmentepithels; C: zystoides Makulaödem und Abhebung der inneren Netzhautschichten in der Fovea. Entspricht dem zystoiden Makulaödem, das im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt wird.
Die Symptome der RP verändern sich je nach Krankheitsstadium. Da die Stäbchenzellen zuerst degenerieren, tritt als frühestes Symptom die Nachtblindheit auf.
Nachtblindheit: verminderte Sehschärfe und Sehschwierigkeiten bei Dunkelheit. Tritt sehr früh auf, da die Stäbchenzellen zuerst degenerieren9); wird im Alter von 10–20 Jahren als Sehschwierigkeit bei schwachem Licht bemerkt. Anfangs ist die Tagessehfunktion oft normal.
Gesichtsfeldeinschränkung: Das Gesichtsfeld verengt sich allmählich von der Peripherie zur Mitte. Entwickelt sich von einem Ringskotom zu einer konzentrischen Gesichtsfeldeinschränkung (Tunnelblick)9).
Sehschärfeminderung: Wenn auf die Stäbchen auch die Zapfendegeneration folgt, nimmt auch die zentrale Sehschärfe ab. Bei Vorliegen eines zystoiden Makulaödems (CME) kann es relativ früh zu einer mäßigen Sehschärfeminderung kommen. In manchen Fällen bleibt die zentrale Sehschärfe bis zum Endstadium erhalten.
Photophobie (Tagblindheit): Überempfindlichkeit gegenüber Licht. Ausdruck einer Zapfenfunktionsstörung. Nimmt mit fortschreitender Zapfendegeneration zu. Wichtig ist die Abgrenzung zur Lichtstreuung durch Katarakt.
Photopsien: können infolge der Degeneration und des Verlusts von Photorezeptoren auftreten.
Visuelle Halluzinationen (Charles-Bonnet-Syndrom) : Bei Patienten mit fortgeschrittener Sehverschlechterung tritt das Phänomen auf, dass sie Landschaften oder Personen sehen, die nicht wirklich existieren. Es handelt sich nicht um eine krankhafte Erfahrung, sondern um ein Phänomen, das durch Überaktivität des visuellen Kortex verursacht wird 9)
Im Folgenden wird ein Überblick über das Fortschreiten der Symptome nach Krankheitsstadium gegeben.
Die klassische Trias der RP ist wie folgt bekannt.
Knochenkörperchenartige Pigmentierung : charakteristische Pigmentierung (bone spicule pattern), die von der mittleren Peripherie zur Peripherie auftritt.
Verengung der Netzhautarterien : sekundär infolge der Photorezeptordegeneration.
Wachsartige Blässe der Papille : spiegelt die Degeneration des Sehnervs wider.
Klassifikation der Krankheitstypen : unterteilt in typische und atypische Formen9).
Typische RP (Stäbchen-Zapfen-Dystrophie) : Stäbchen werden zuerst geschädigt, Zapfen später.
Stäbchendystrophie (Subtyp) : Zapfen bleiben bis zum Endstadium unbeeinträchtigt; zentrale Sehschärfe bleibt auch bei schwerer konzentrischer Gesichtsfeldeinschränkung erhalten.
Atypische RP9) :
Pigmentlose RP : keine Pigmentierung nachweisbar.
Einseitige RP : nur auf einem Auge oder mit starkem Seitenunterschied.
Sektorielle RP : auf 1–2 Quadranten der Netzhaut beschränkt; langsame Progression, gute Prognose.
Zentrale/parazentrale RP : Netzhautläsionen und Gesichtsfeldausfälle beginnen zentral.
Retinitis punctata albescens : weiße bis gelbe punktförmige Läsionen in der Netzhaut.
Bei Kindern fehlen oft die typischen Befunde, und das ERG ist der Schlüssel zur Diagnose.
Hintere subkapsuläre Katarakt (HSK) : tritt bei etwa 50 % auf. Charakteristisch ist eine Sehverschlechterung bei Helligkeit. Eine EZ (Ellipsoidzone)-Breite ≥ 600 μm sagt eine gute postoperative Sehschärfe voraus (AUC 0,97) 5).
Zystoides Makulaödem (CME) : tritt bei 10–50 % auf und ist die Hauptursache für zentrale Sehverschlechterung 9).
Winkelblockglaukom : bei etwa 1 % wurden Anfälle berichtet; aufgrund der Schwäche der Zonulafasern kann auch eine Linsensubluxation auftreten 9).
Makulaloch / foveales Schisis : relativ selten, kann aber eine Vitrektomie erforderlich machen 9).
QVerbessert eine Kataraktoperation die Sehkraft?
A
Bei der Kataraktoperation der hinteren subkapsulären Katarakt in Verbindung mit RP ist bei einer präoperativen OCT-EZ-Breite ≥ 600 μm eine gute postoperative Sehschärfe zu erwarten (AUC 0,97) 5). Die EZ-Breite ist ein nützlicher Biomarker zur präoperativen Vorhersage der Sehfunktion. Allerdings sind die Zonulafasern oft schwach, sodass auf eine vordere Kapselkontraktur und IOL-Dislokation geachtet werden muss. Zur Prophylaxe eines postoperativen CME (10–14 %) wird eine längere als übliche Anwendung von Steroid- und NSAR-Augentropfen empfohlen 9).
RP ist eine genetisch heterogene Gruppe von Erkrankungen, die durch Mutationen in über 100 Genen verursacht wird 9). Die wichtigsten verursachenden Gene bei Japanern sind nach Vererbungsmodus aufgeführt.
Vergleich der wichtigsten verursachenden Gene unten.
Gen
Vererbung
Häufigkeit/Merkmale bei Japanern
EYS
AR
30–50 % der Fälle mit identifiziertem Gen (häufigster bei AR-Typ) 12)13)
USH2A
AR
Zweithäufigster bei AR-Form (4-9 %); Hauptgen des Usher-Syndroms 12)
RHO
AD
Häufigster bei AD-Form 6)
RPGR
X-chromosomal
Etwa 70-75 % bei XL-Form 6)
REEP6
AR
Eines der ursächlichen Gene der AR-Form 4)
Die Merkmale der einzelnen Gene werden im Folgenden ergänzt.
EYS (Eyes Shut Homolog) : Häufigstes ursächliches Gen der AR-RP bei Japanern (30-50 % der identifizierten Fälle) 12)13). In westlichen Ländern ist es nicht so häufig, was den spezifischen genetischen Hintergrund der Japaner widerspiegelt.
USH2A : Hauptgen des Usher-Syndroms (RP + Hörverlust) und zweithäufigstes bei japanischer AR-RP nach EYS (4-9 %) 12)
RHO (Rhodopsin) : häufigstes ursächliches Gen für AD-RP6). Es kodiert für das Photorezeptorprotein der Stäbchen.
RPGR (Retinitis Pigmentosa GTPase Regulator) : Hauptursache für XL-RP6). Bei männlichen Patienten mit RPGR-Mutationen wurden Fälle von primärer Ziliendysfunktion (PCD) berichtet1).
REEP6 (Receptor Expression-Enhancing Protein 6) : eines der ursächlichen Gene für AR-RP4).
Die Nachweisrate ursächlicher Gene durch Gentests variiert je nach Vererbungsmuster. Sie wird für AD mit 35–60 %, für AR und sporadische Fälle mit 30–50 % und für XL mit 16–36 % angegeben6).
Darüber hinaus treten bei syndromaler RP, einschließlich Joubert-Syndrom, Bardet-Biedl-Syndrom usw., häufig Mutationen in zilienassoziierten Genen auf, die mit systemischen Komplikationen (Nierenerkrankungen, Polydaktylie, Adipositas usw.) einhergehen können2). Die Abgrenzung zu verschiedenen Syndromen wie Friedreich-Ataxie (PHARC), PCARP, Oliver-McFarlane-Syndrom usw. ist ebenfalls wichtig3).
QSollte man einen Gentest durchführen lassen?
A
Die genetische Diagnostik ist wichtig für die endgültige Diagnose, die genetische Beratung und die Bestimmung der Eignung für eine Gentherapie. Das PrismGuide IRD-Panel-System (umfassende Analyse der Exonsequenzen von 82 IRD-ursächlichen Genen) wurde 2023 von der Krankenkasse übernommen, aber Stand Juni 2025 sind nur junge Patienten mit Verdacht auf RPE65-bedingte IRD anspruchsberechtigt9). Die Durchführung in Kombination mit einer genetischen Beratung wird empfohlen. Eine genetische Beratung kann auch ohne genetische Diagnostik in Anspruch genommen werden.
Abnormaler hyperfluoreszierender Ring (AF-Ring) als Stadiumsindikator 6)
Gesichtsfelduntersuchung
Progressionsbewertung
Goldmann-Perimeter; HFA 10-2 Programm ebenfalls nützlich 9)10)
Farbsehprüfung
Bewertung der Zapfenfunktion
Erworbene Blau-Gelb-Anomalie häufig; Panel D-15 und 100-Hue-Test 9)
Dunkeladaptationsprüfung
Bewertung der Stäbchenfunktion
Kohlrausch-Knickpunkt nicht nachweisbar 10)
NGS-Gentest
Genetische Diagnostik
PrismGuide IRD-Panel (82 Gene) 9)
Die Details der einzelnen Untersuchungen sind im Folgenden aufgeführt.
Elektroretinographie (ERG) : Für die definitive Diagnose unerlässlich 6)9). Die Stäbchenantwort (skotopisches ERG) ist bereits früh vermindert, und mit Fortschreiten nimmt auch die Zapfenantwort (photopisches ERG) ab. Das Ganzfeld-ERG ist die Grundlage. Zum Zeitpunkt der Vorstellung ist es oft bereits nicht mehr ableitbar.
OCT (Optische Kohärenztomographie) : Beurteilung der Breite der Ellipsoidzone (EZ) und des Verschwindensmusters. Die EZ-Breite ist ein nützlicher quantitativer Biomarker für Sehfunktion und Prognose und wird auch zur Entscheidung über die Indikation einer Kataraktoperation herangezogen 5). Bereits im Frühstadium zeigen sich eine Ausdünnung der äußeren Körnerschicht und ein Verlust der EZ.
Fundusautofluoreszenz (FAF) : Ein abnormaler hyperfluoreszenter Ring (autofluoreszenter Ring; AF-Ring) erscheint um die Makula, der die verbleibende funktionelle Netzhaut anzeigt und als Indikator für das Krankheitsstadium dient 6).
Gesichtsfelduntersuchung : Die dynamische Perimetrie mit dem Goldmann-Perimeter ist der Standard. Mit Fortschreiten kommt es zu einem Ringskotom und dann zu einer konzentrischen Gesichtsfeldeinschränkung 10). Das Humphrey-Gesichtsfeld (HFA) 10-2-Programm ist nützlich zur Beurteilung der verbleibenden zentralen Zapfenfunktion 9).
Farbsehprüfung : Eine erworbene Blau-Gelb-Farbenstörung wird häufig beobachtet. Die Beurteilung erfolgt mit dem Panel D-15-Test und dem 100-Hue-Test 9).
Dunkeladaptationstest : Der Kohlrausch-Knickpunkt (Umschaltpunkt zwischen Stäbchen und Zapfen) wird nicht nachgewiesen 10).
Next-Generation-Sequencing (NGS) : Mit dem PrismGuide IRD-Panel-System können die Exonsequenzen von 82 krankheitsverursachenden Genen umfassend analysiert werden 9). Es ist auch für die Indikationsstellung zur Gentherapie unverzichtbar.
Derzeit gibt es keine Heilung für RP6)9). Die Behandlung konzentriert sich auf den Erhalt der Sehfunktion, die Behandlung von Komplikationen und die Unterstützung des sozialen Lebens.
Schutz der Photorezeptoren
Vitamin A (15.000 IE/Tag) : Orale Gabe soll die Verschlechterung des ERG um einige Prozent verzögern14). Keine Verbesserung von Sehschärfe oder Gesichtsfeld. Bei Langzeiteinnahme ist eine Überwachung der Leberfunktion erforderlich. In der Schwangerschaft aufgrund teratogener Wirkung kontraindiziert. Bei ABCA4-Mutation kann es das Fortschreiten fördern14). Vitamin A kann das Fortschreiten beschleunigen, daher ist Vorsicht geboten14).
Unoproston-Augentropfen : Es wurde eine dosisabhängige Tendenz zur Verbesserung der Empfindlichkeit beobachtet, aber der primäre Endpunkt der Phase-2-Studie (zentrale 2-Grad-Netzhautempfindlichkeit) zeigte keinen signifikanten Unterschied16).
Nilvadipin (Kalziumantagonist) : Ein Langzeitbericht zeigte eine Verlangsamung des Fortschreitens von Gesichtsfeldausfällen15). Es handelt sich um einen Einzelzentrumsbericht mit wenigen Fällen; multizentrische Nachuntersuchungen wurden nicht durchgeführt.
N-Acetylcystein (NAC) : Unterdrückung von oxidativem Stress. Eine Phase-I-Studie berichtete über eine Verbesserung der Sehschärfe17), und 2025 läuft eine Phase-III-Studie.
DHA und Lutein : Schützen die Makula-Photorezeptoren vor oxidativem Stress. Ein zusätzlicher Effekt von DHA zu Vitamin A wurde nicht festgestellt.
Helenien (Adaptinol) : Zugelassen zur vorübergehenden Verbesserung von Gesichtsfeld und Dunkeladaptation bei RP. Eine Wirksamkeitsbewertung nach modernen medizinischen Standards wurde nicht durchgeführt.
Lichtschutzbrille : Reduziert oxidativen Stress durch UV- und helles Licht. Tägliche Anwendung wird empfohlen.
Die erste Wahl ist ein Carboanhydrasehemmer (CAI). Verwendung von Dorzolamid (Trusopt) Augentropfen oder Acetazolamid (Diamox) oral. Eine Verbesserung der zentralen Makuladicke wird in etwa 40 % erreicht. Bei etwa 30 % tritt ein Rezidiv auf9).
Bei CAI-Resistenz sind Steroide in Betracht zu ziehen. Intravitreale Injektion von Triamcinolonacetonid (Maquaid) oder Dexamethason-Implantat (Ozurdex).
Anti-VEGF-Medikamente werden bei RP-CME nicht empfohlen, da die VEGF-Produktion reduziert ist9).
Beachten Sie, dass keines dieser Verfahren für RP-CME zugelassen ist und es sich um Off-Label-Anwendungen handelt.
Kataraktoperation: Durchgeführt bei hinterer subkapsulärer Katarakt. Eine präoperative OCT-EZ-Breite ≥ 600 μm ist ein prädiktiver Faktor für eine gute postoperative Sehschärfe5). Bei Zonulafragilität sollte die Verwendung eines Kapselspannrings erwogen werden. Zur Vorbeugung eines postoperativen CME (10–14 %) werden Steroid- und NSAR-Augentropfen länger als üblich angewendet 9).
Winkelblockglaukom: Das Risiko eines primären Winkelblockglaukoms ist bei RP-Patienten erhöht. Die Vorderkammer wird allmählich flacher; prophylaktisch wird eine Laser-Iridotomie oder Kataraktoperation durchgeführt 9).
Epiretinale Membran (GL2026 CQ4): Vitrektomie. Bei kontinuierlicher EZ-Linie ist eine Verbesserung der Sehschärfe zu erwarten. Bei diskontinuierlicher EZ-Linie ist die Erholung begrenzt. Es wurde über langfristige schwere Makulaatrophie berichtet; eine Untersuchung in einem spezialisierten Zentrum ist wünschenswert 9).
Makulaloch: Die Vitrektomie ist die einzige kurative Behandlung. Die postoperativen Ergebnisse sind nur begrenzt untersucht 9).
Unterstützung und Rehabilitation
Low-Vision-Versorgung: Sehschwäche → Lupen, Bildschirmlesegeräte, Tablets; Photophobie → getönte Brillen; Gesichtsfeldeinschränkung → weißer Stock; Fernsicht → Monokular; Nachtsichthilfsbrillen. Eine individuelle Unterstützung entsprechend Gesichtsfeld und Sehschärfe ist wichtig. Die Nutzung von Smart Site (Vorstellung von Low-Vision-Beratungsstellen in den jeweiligen Regionen) wird empfohlen.
Genetische Beratung: Durchgeführt von einem klinischen Genetiker und einem zertifizierten genetischen Berater. Häufige Themen sind die Einschätzung des Wiederholungsrisikos, Ausbildung, Beruf, Heirat und Kinderwunsch. Eine genetische Beratung ist auch ohne vorherige genetische Diagnostik möglich.
System für seltene Krankheiten: Als designierte seltene Krankheit ist das System zur Kostenübernahme medizinischer Behandlungen nutzbar 9). Auch die Beantragung eines Schwerbehindertenausweises und einer medizinischen Unterstützung zur Selbstständigkeit sollte erwogen werden.
Voretigen Neparvovec (Luxturna): Gentherapeutikum, das bei Patienten mit biallelischen pathogenen Varianten des RPE65-Gens und ausreichend lebensfähigen Netzhautzellen verabreicht werden kann. 2023 in Japan zugelassen 9). In der US-amerikanischen Phase-III-Studie (301) wurden 31 Patienten eingeschlossen; die mITT-Analyse (Intervention 20, Kontrolle 9) zeigte eine signifikante Verbesserung von MLMT und Weißlicht-FST gegenüber der Kontrollgruppe 18). In der nationalen Phase-III-Studie (A11301) wurde bei 4 japanischen Patienten eine erhöhte FST-Empfindlichkeit und Gesichtsfeldausweitung bestätigt 19). Die Verbesserung der Sehschärfe ist gering, und bei über 20 % wurde als Langzeitkomplikation eine Netzhaut-Aderhaut-Atrophie berichtet 9).
Alle 6 Monate bis 1 Jahr durchführen: Sehschärfe, Spaltlampenmikroskopie, Funduskopie, Humphrey-Gesichtsfeld (HFA 10-2), OCT9).
QWelche Medikamente werden zur Behandlung des Makulaödems eingesetzt?
A
Die Erstlinientherapie des RP-CME sind Carboanhydrasehemmer (CAH), die als Dorzolamid-Augentropfen oder orales Acetazolamid verabreicht werden 9). Eine Verbesserung der zentralen Makuladicke wird in etwa 40 % der Fälle erreicht, aber bei etwa 30 % tritt ein Rezidiv auf. Bei fehlender Besserung unter CAH kommen intravitreale Triamcinolonacetonid-Injektionen oder ein intravitreales Dexamethason-Implantat (Ozurdex) in Frage. Anti-VEGF-Medikamente werden bei RP-CME nicht empfohlen. Es ist zu beachten, dass alle diese Anwendungen off-label erfolgen.
6. Pathophysiologie und detaillierte Krankheitsmechanismen
Der letzte gemeinsame Weg des Photorezeptor-Zelltods bei RP ist die Apoptose. Obwohl die Arten von Genmutationen vielfältig sind, konvergieren sie letztlich auf einen gemeinsamen Zelltodweg.
Bei RP ist das typische Muster, dass zuerst die Stäbchenphotorezeptoren degenerieren und verschwinden, gefolgt von einer sekundären Degeneration der Zapfen7). Es wird angenommen, dass Zapfen von einem von Stäbchen produzierten Überlebensfaktor (RdCVF: Rod-derived Cone Viability Factor) abhängig sind; daher verlieren Zapfen nach dem Verlust der Stäbchen ebenfalls ihre Funktion 7)11).
Die Netzhaut ist eines der stoffwechselaktivsten Gewebe; sie wandelt 80–90 % der Glukose durch aerobe Glykolyse (Warburg-Effekt) in Laktat um. Zapfen sind anfälliger für metabolischen Stress als Stäbchen, und diese metabolische Vulnerabilität trägt ebenfalls zur sekundären Zapfendegeneration bei 11).
Entzündung wird ebenfalls als Hauptfaktor für das Fortschreiten der RP erkannt; die Aktivierung von Mikroglia und die Infiltration von Makrophagen verschlimmern die Netzhautschädigung 11). Oxidativer Stress wirkt ebenfalls als biologischer Treiber der sekundären Zapfendegeneration.
REEP6-Mutation : REEP6 kodiert für ein Protein, das an der Aufrechterhaltung der ER-Morphologie beteiligt ist. Pathogene Mutationen führen zur Bildung von ER-Einschlusskörpern im äußeren Segment der Stäbchen, was zur Photorezeptordegeneration führt4)
RPGR-Mutation : RPGR ist an der Axonemstruktur primärer Zilien beteiligt; Mutationen beeinträchtigen den Transport von Substanzen zum äußeren Segment der Photorezeptoren1)
7. Aktuelle Forschung und Zukunftsaussichten (Berichte aus der Forschungsphase)
Die Gentherapie ist der vielversprechendste Ansatz zur Behandlung erblicher Netzhauterkrankungen8).
Luxturna (Voretigen Neparvovec) : Gentherapeutikum, das Patienten mit biallelischen pathogenen Varianten des RPE65-Gens verabreicht werden kann. In der US-amerikanischen Phase-III-Studie (301-Studie) wurden 31 Patienten eingeschlossen; die mITT-Analyse (20 Intervention, 9 Kontrolle) zeigte eine signifikante Verbesserung des MLMT und des Weißlicht-FST gegenüber der Kontrollgruppe18). In der nationalen Phase-III-Studie (A11301-Studie) wurde bei 4 japanischen Patienten eine Erhöhung der FST-Empfindlichkeit und eine Gesichtsfeldausweitung bestätigt19). 2023 in Japan zugelassen, stellt es eine Brücke zwischen Standardbehandlung und experimenteller Therapie dar.
RPGR-Gentherapie : Die AAV-vermittelte Gentherapie für XL-RP aufgrund von RPGR-Mutationen hat klinische Studien der Phase I/II/III erreicht8).
CRISPR/Cas9 : Die Forschung zur direkten Korrektur pathogener Mutationen oder zur Inaktivierung dominanter negativer Mutationen ist im Gange8).
RdCVF (Stäbchen-abgeleiteter Zapfen-Überlebensfaktor) und Zapfenschutztherapie
RdCVF ist ein Protein, das von Stäbchen sezerniert wird und das Überleben von Zapfen aufrechterhält7)11). Klinische Studien zur Zapfenschutztherapie mit RdCVF laufen, und sie wird als unabhängige Behandlungsstrategie zur Erhaltung der Zapfenfunktion nach Stäbchendegeneration angesehen.
NAC ist ein Medikament, das oxidativen Stress unterdrückt; eine Phase-I-Studie berichtete über eine Verbesserung der Sehschärfe17). Im Jahr 2025 läuft eine Phase-III-Studie.
Umwidmungspotenzial von Glukokortikoiden (Dexamethason)
Aktuelle In-vivo-Studien (rd10-Mausmodell) haben gezeigt, dass intravitreales Dexamethason die Zapfen-Photorezeptoren und das retinale Pigmentepithel schützt 11). Glukokortikoide haben ein starkes Potenzial für die Umwidmung als mutationsunabhängige Therapie. Allerdings beschränken sich die derzeitigen Belege auf Tiermodelle, und für eine klinische Anwendung beim Menschen sind weitere Validierungen erforderlich.
iPS-Zell-abgeleitete Netzhauttransplantation und künstliche Netzhaut
iPS-Zell-abgeleitete Netzhauttransplantation: Die Forschung zur Transplantation von Photorezeptor-Blättchen, die aus patienteneigenen iPS-Zellen hergestellt wurden, schreitet voran.
Künstliche Netzhaut (Netzhautprothese): Elektrisches Stimulationsgerät für terminale RP. Argus II und andere sind im Ausland kommerziell erhältlich, und in Japan laufen klinische Studien zur suprachoroidalen transretinalen Stimulation.
QIst Gentherapie in Japan verfügbar?
A
Voretigen Neparvovec (Luxturna) wurde 2023 in Japan zugelassen, jedoch nur für Netzhautdystrophien mit biallelischen pathogenen Varianten des RPE65-Gens 9)18)19). Die Gentherapie für RP mit anderen Genmutationen, einschließlich RPGR-Mutationen, befindet sich noch in der klinischen Erprobung 7) und ist in Japan nicht als allgemeine Behandlung zugelassen.
QWie erhält man Zugang zu experimentellen Behandlungen?
A
Die Teilnahme an klinischen Studien ist auf offizielle Studien beschränkt, die von der Ethikkommission der medizinischen Einrichtung genehmigt wurden. Neben der Konsultation des behandelnden Arztes können Studieninformationen auf der vom Nationalen Krebszentrum betriebenen klinischen Studieninformationsseite (jRCT) oder auf der US-amerikanischen Website clinicaltrials.gov gesucht werden.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.