La rétinite pigmentaire (RP) est un terme générique désignant un groupe de maladies héréditaires caractérisées par une dégénérescence progressive et étendue des photorécepteurs (bâtonnets et cônes) et de l’épithélium pigmentaire rétinien (EPR). La dégénérescence des bâtonnets précède celle des cônes ; on parle alors de dystrophie des bâtonnets et des cônes, et la RP est considérée comme synonyme. Il ne s’agit pas d’une maladie unique mais d’un groupe de maladies impliquant plus de 100 gènes.
La prévalence est de 1 personne sur 4 000 à 8 000, et le nombre total de patients au Japon dépasse au moins 30 000 (20 687 bénéficiaires de la maladie rare désignée en 2023) 9). En ce qui concerne la déficience visuelle, elle est la deuxième cause de déficience visuelle (13,0 %) chez les nouveaux titulaires d’un certificat de handicap visuel âgés de 18 ans et plus (après le glaucome, 40,7 % en 2019), et la première cause de cécité congénitale 9). Au Japon, elle est reconnue comme maladie rare désignée en vertu de la loi sur les maladies rares (depuis le 1er janvier 2015) 9) et donne droit à une aide financière pour les frais médicaux.
Il existe également des RP syndromiques associés à d’autres maladies systémiques, classés sous le concept général de ciliopathie comme suit 9)2).
Ciliopathie :
Syndrome d’Usher (type 1/2/3) : RP + surdité ; maladie désignée (AR). Le type 1 s’accompagne d’une surdité sévère et de troubles vestibulaires dès la petite enfance.
Mucopolysaccharidose (Hurler, Hunter) : avec opacité du fond d’œil
Maladie de Refsum (type adulte et infantile) : maladie peroxysomale ; ataxie cérébelleuse, neuropathie périphérique (AR)
Syndrome de Bassen-Kornzweig : trouble du métabolisme lipidique
Maladie mitochondriale :
Syndrome de Kearns-Sayre : ophtalmoplégie externe progressive bilatérale, ptosis, trouble de la conduction cardiaque
Dystrophie musculaire :
Dystrophie myotonique : peut être associée à une RP
De plus, il est important de différencier la RP de divers syndromes tels que PHARC (neuropathie périphérique, surdité, ataxie, RP, cataracte), PCARP, et le syndrome d’Oliver-McFarlane3).
QLa rétinite pigmentaire est-elle héréditaire ?
A
La RP est une maladie génétique, mais elle n’est pas nécessairement transmise à tous les enfants. Le risque de transmission à l’enfant varie selon le mode de transmission. Dans la forme autosomique dominante (AD), il y a 50 % de risque de transmission à l’enfant, mais dans les formes autosomique récessive (AR) ou liée à l’X (XL), le risque varie selon le mode de transmission. Dans les cas sporadiques (48 à 63 % des cas), le risque de transmission à la génération suivante est souvent relativement faible9). Le recours au conseil génétique est recommandé.
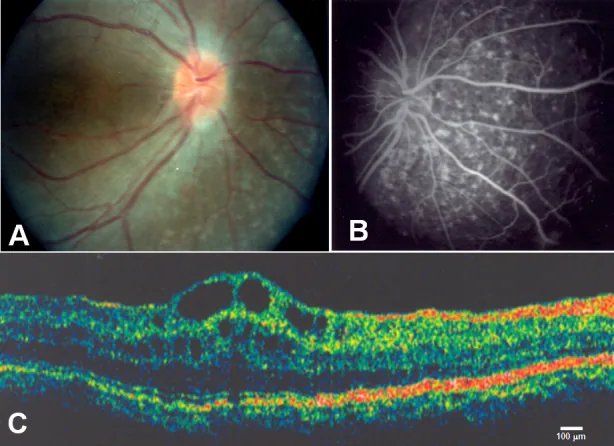
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A : drusen de la papille optique et atrophie étendue de l’épithélium pigmentaire rétinien ; B : hyperfluorescence choroïdienne correspondant à l’atrophie de l’épithélium pigmentaire rétinien ; C : œdème maculaire cystoïde et décollement des couches rétiniennes internes au niveau de la fovéa. Correspond à l’œdème maculaire cystoïde traité dans la section « 2. Principaux symptômes et signes cliniques ».
Les symptômes de la RP évoluent selon le stade de la maladie. Les bâtonnets dégénérant en premier, la cécité nocturne est le symptôme le plus précoce.
Cécité nocturne : baisse de l’acuité visuelle et difficulté à voir dans l’obscurité. Apparaît très tôt car les bâtonnets dégénèrent en premier9) ; elle est ressentie entre 10 et 20 ans comme une difficulté à voir dans les endroits faiblement éclairés. Au début, la fonction visuelle diurne est souvent normale.
Rétrécissement du champ visuel : le champ visuel se rétrécit progressivement de la périphérie vers le centre. Évolue d’un scotome annulaire vers un rétrécissement concentrique (vision en tunnel)9).
Baisse de l’acuité visuelle : lorsque la dégénérescence des cônes suit celle des bâtonnets, l’acuité visuelle centrale diminue également. En cas d’œdème maculaire cystoïde (CME), une baisse modérée de l’acuité visuelle peut survenir relativement tôt. Dans certains cas, l’acuité visuelle centrale peut être préservée jusqu’à un stade avancé.
Photophobie (cécité diurne) : sensibilité à la lumière. Manifestation d’un dysfonctionnement des cônes. S’aggrave avec la progression de la dégénérescence des cônes. Important de différencier de la diffusion de la lumière due à la cataracte.
Phosphènes : peuvent survenir en raison de la dégénérescence et de la perte des photorécepteurs.
Hallucinations visuelles (syndrome de Charles Bonnet) : Chez les patients dont l’acuité visuelle a diminué, phénomène où ils voient des paysages ou des personnes qui n’existent pas réellement. Il ne s’agit pas d’une expérience pathologique, mais d’un phénomène dû à une hyperactivité du cortex visuel 9)
Voici un aperçu de la progression des symptômes par stade de la maladie.
Stade
Principaux symptômes
Période indicative
Début
Cécité nocturne
10-20 ans
Intermédiaire
Rétrécissement du champ visuel (scotome annulaire → concentrique)
30-40 ans
Tardif
Baisse de l’acuité visuelle, anomalies de la vision des couleurs, photophobie
50 ans et plus
Signes cliniques et classification des types de maladie
La triade classique de la RP est connue comme suit.
Dépôts pigmentaires en spicules osseux : pigmentation caractéristique (motif en spicules osseux) apparaissant de la région périphérique moyenne à la périphérie.
Rétrécissement des artères rétiniennes : secondaire à la dégénérescence des photorécepteurs.
Pâleur cireuse de la papille optique : reflète la dégénérescence du nerf optique.
Classification des types : on distingue les formes typiques et atypiques9).
RP typique (dystrophie des bâtonnets et des cônes) : les bâtonnets sont touchés en premier, puis les cônes.
Dystrophie des bâtonnets (sous-type) : les cônes ne sont pas touchés jusqu’aux stades avancés ; l’acuité visuelle centrale est préservée même en cas de rétrécissement concentrique sévère du champ visuel.
RP atypique9) :
RP sans pigment : absence de dépôts pigmentaires.
RP unilatérale : atteinte d’un seul œil ou asymétrie marquée.
RP sectorielle : limitée à 1 à 2 quadrants de la rétine ; progression lente, bon pronostic.
RP centrale ou paracentrale : lésions rétiniennes et anomalies du champ visuel débutant au centre.
Rétinopathie ponctuée blanche : lésions ponctuées blanches à jaunes dans la rétine.
Chez l’enfant, les signes typiques sont souvent absents, et l’ERG est la clé du diagnostic.
Cataracte sous-capsulaire postérieure (CSP) : survient dans environ 50 % des cas. Caractérisée par une baisse de l’acuité visuelle en lumière vive. Une largeur de la zone ellipsoïde (EZ) ≥ 600 μm prédit une bonne acuité visuelle postopératoire (AUC 0,97) 5).
Œdème maculaire cystoïde (OMC) : survient dans 10 à 50 % des cas et constitue la principale cause de baisse de l’acuité visuelle centrale 9).
Glaucome par fermeture de l’angle : des crises sont rapportées dans environ 1 % des cas ; une subluxation du cristallin due à une fragilisation des zonules de Zinn peut également survenir 9).
Trou maculaire / schisis fovéolaire : relativement rare, mais peut nécessiter une vitrectomie9).
QLa chirurgie de la cataracte améliore-t-elle la vision ?
A
Pour la chirurgie de la cataracte sous-capsulaire postérieure associée à la RP, si la largeur de la zone ellipsoïde (EZ) à l’OCT préopératoire est ≥ 600 μm, une bonne acuité visuelle postopératoire est attendue (AUC 0,97) 5). La largeur de l’EZ est un biomarqueur utile pour prédire la fonction visuelle préopératoire. Cependant, les zonules de Zinn sont souvent fragiles, nécessitant une attention particulière à la contraction capsulaire antérieure et à la luxation du cristallin artificiel. Pour prévenir l’OMC postopératoire (10-14 %), une utilisation prolongée de collyres stéroïdiens et AINS est recommandée 9).
La RP est un groupe de maladies très hétérogènes sur le plan génétique, causées par des mutations de plus de 100 gènes 9). Les principaux gènes responsables chez les Japonais sont présentés par mode de transmission.
Comparaison des principaux gènes responsables ci-dessous.
Gène
Mode de transmission
Fréquence/caractéristiques chez les Japonais
EYS
AR
30-50 % des cas avec gène identifié (le plus fréquent dans la forme AR) 12)13)
USH2A
AR
Deuxième plus fréquent dans la forme AR (4-9 %) ; gène majeur du syndrome d’Usher 12)
RHO
AD
Le plus fréquent dans la forme AD 6)
RPGR
Lié à l’X
Environ 70-75 % des cas de forme XL 6)
REEP6
AR
Un des gènes responsables de la forme AR 4)
Les caractéristiques de chaque gène sont détaillées ci-dessous.
EYS (Eyes Shut Homolog) : Gène le plus fréquent de la RP de forme AR chez les Japonais (30-50 % des cas identifiés) 12)13). Sa fréquence est beaucoup plus faible en Occident, reflétant un contexte génétique propre aux Japonais.
USH2A : Gène majeur du syndrome d’Usher (RP + surdité), deuxième plus fréquent dans la RP de forme AR chez les Japonais après EYS (4-9 %) 12)
RHO (Rhodopsine) : le gène le plus fréquemment responsable de la RP de type AD6). Il code pour la protéine photoréceptrice des bâtonnets.
RPGR (Retinitis Pigmentosa GTPase Regulator) : principal gène responsable de la RP de type XL6). Des cas de dysfonction ciliaire primitive (PCD) ont été rapportés chez des patients masculins porteurs de mutations de RPGR1).
REEP6 (Receptor Expression-Enhancing Protein 6) : l’un des gènes responsables de la RP de type AR4).
Le taux de détection des gènes responsables par les tests génétiques varie selon le mode de transmission. Il est estimé à 35-60 % pour le type AD, 30-50 % pour le type AR et les cas sporadiques, et 16-36 % pour le type XL6).
De plus, dans la RP syndromique, incluant le syndrome de Joubert, le syndrome de Bardet-Biedl, etc., les mutations des gènes liés aux cils sont fréquentes et peuvent s’accompagner de complications systémiques (maladies rénales, polydactylie, obésité, etc.)2). Il est également important de différencier la RP de divers syndromes tels que l’ataxie de Friedreich (PHARC), la PCARP, le syndrome d’Oliver-McFarlane, etc.3).
QFaut-il subir un test génétique ?
A
Le diagnostic génétique est important pour le diagnostic définitif, le conseil génétique et la détermination de l’éligibilité à la thérapie génique. Le système PrismGuide IRD Panel (analyse complète des séquences exoniques de 82 gènes pathogènes de la DRS) a été approuvé par l’assurance maladie en 2023, mais en juin 2025, seuls les patients jeunes suspectés de DRS liée à RPE65 sont éligibles9). Il est recommandé de le réaliser en combinaison avec un conseil génétique. Il est possible de bénéficier d’un conseil génétique sans avoir subi de diagnostic génétique.
Anneau hyperfluorescent anormal (AF ring) comme indicateur de stade 6)
Examen du champ visuel
Évaluation de la progression
Périmètre de Goldmann ; le programme HFA 10-2 est également utile 9)10)
Examen de la vision des couleurs
Évaluation de la fonction des cônes
Dyschromatopsie bleu-jaune acquise fréquente ; test Panel D-15 et 100 Hue 9)
Examen d’adaptation à l’obscurité
Évaluation de la fonction des bâtonnets
Absence de point d’inflexion de Kohlrausch 10)
Test génétique NGS
Diagnostic génétique
Panel PrismGuide IRD (82 gènes) 9)
Les détails de chaque examen sont présentés ci-dessous.
Électrorétinographie (ERG) : Essentielle pour le diagnostic définitif 6)9). La réponse des bâtonnets (ERG scotopique) diminue dès le début, et la réponse des cônes (ERG photopique) diminue également avec la progression. L’ERG plein champ est la base. Il arrive souvent que l’ERG soit déjà non enregistrable lors de la consultation.
OCT (tomographie par cohérence optique) : Évalue la largeur de la zone ellipsoïde (EZ) et le schéma de disparition. La largeur de l’EZ est utile comme biomarqueur quantitatif de la fonction visuelle et du pronostic, et est également utilisée pour décider de l’indication de la chirurgie de la cataracte5). Dès le début de la maladie, on observe un amincissement de la couche nucléaire externe et une disparition de l’EZ.
Autofluorescence du fond d’œil (FAF) : Un anneau hyperfluorescent anormal (anneau autofluorescent ; AF ring) apparaît autour de la macula, indiquant la rétine fonctionnelle restante et servant d’indicateur de progression de la maladie 6).
Examen du champ visuel : La périmétrie dynamique avec le périmètre de Goldmann est la norme. Avec la progression, on observe un scotome annulaire puis un rétrécissement concentrique du champ visuel 10). Le programme 10-2 du périmètre de Humphrey (HFA) est utile pour évaluer la fonction résiduelle des cônes centraux 9).
Examen de la vision des couleurs : Une dyschromatopsie bleu-jaune acquise est fréquemment observée. Elle est évaluée par le test Panel D-15 et le test 100 Hue 9).
Examen d’adaptation à l’obscurité : Le point d’inflexion de Kohlrausch (point de transition entre bâtonnets et cônes) n’est pas détecté 10).
Séquençage de nouvelle génération (NGS) : Le système de panel PrismGuide IRD permet d’analyser de manière exhaustive les séquences exoniques de 82 gènes pathogènes 9). Il est également indispensable pour déterminer l’indication de la thérapie génique.
À l’heure actuelle, il n’existe pas de traitement curatif de la RP6)9). Le traitement se concentre sur le maintien de la fonction visuelle, la gestion des complications et le soutien à la vie sociale.
Protection des photorécepteurs
Vitamine A (15 000 UI/jour) : Des rapports indiquent que l’administration orale retarde la détérioration de l’ERG de quelques pour cent14). Aucun effet d’amélioration de l’acuité visuelle ou du champ visuel. Une surveillance de la fonction hépatique est nécessaire en cas d’utilisation à long terme. Contre-indiqué pendant la grossesse en raison d’un risque tératogène. Peut favoriser la progression en cas de mutation ABCA414). La vitamine E peut accélérer la progression et nécessite une attention particulière14).
Collyre d’unoprostone : Une tendance à l’amélioration de la sensibilité dose-dépendante a été observée, mais le critère principal de l’essai de phase 2 (sensibilité rétinienne centrale à 2 degrés) n’a pas montré de différence significative16).
Nilvadipine (antagoniste calcique) : Un rapport à long terme a montré un ralentissement de la progression des défauts du champ visuel15). Il s’agit d’un rapport monocentrique avec un petit nombre de cas, et aucune étude multicentrique de suivi n’a été réalisée.
N-acétylcystéine (NAC) : Suppression du stress oxydatif. Un essai de phase I a rapporté une amélioration de l’acuité visuelle17), et un essai de phase III est en cours en 2025.
DHA et lutéine : Protègent les photorécepteurs maculaires du stress oxydatif. Aucun effet additif du DHA à la vitamine A n’a été observé.
Hélénien (Adaptinol) : Approuvé pour l’amélioration temporaire du champ visuel et de l’adaptation à l’obscurité dans la RP. Aucune évaluation de l’efficacité selon les normes médicales modernes n’a été réalisée.
Lunettes de protection contre la lumière : Réduisent le stress oxydatif dû aux UV et à la lumière intense. L’utilisation quotidienne est recommandée.
Traitement des complications
Traitement de l’œdème maculaire cystoïde (OMC) :
Le traitement de première intention est un inhibiteur de l’anhydrase carbonique (IAC). Utilisation de collyre de dorzolamide (Trusopt) ou d’acétazolamide (Diamox) par voie orale. Une amélioration de l’épaisseur maculaire centrale est obtenue dans environ 40 % des cas. Une récidive est observée dans environ 30 % des cas9).
Les anti-VEGF ne sont pas recommandés dans l’OMC-RP car la production de VEGF est réduite9).
Notez qu’aucun de ces traitements n’est approuvé pour la RP-CME et qu’ils sont utilisés hors AMM.
Chirurgie de la cataracte : réalisée en cas de cataracte sous-capsulaire postérieure. Une largeur de zone ellipsoïde (EZ) ≥ 600 μm en OCT préopératoire est un facteur prédictif d’une bonne acuité visuelle postopératoire 5). En cas de fragilité des zonules de Zinn, envisager l’utilisation d’un anneau de tension capsulaire. Pour prévenir l’œdème maculaire cystoïde postopératoire (10-14 %), utiliser des collyres stéroïdiens et AINS pendant une période plus longue que d’habitude 9).
Glaucome par fermeture de l’angle : le risque de glaucome primitif par fermeture de l’angle est élevé chez les patients RP. La chambre antérieure devient progressivement peu profonde ; réaliser une iridotomie au laser prophylactique ou une chirurgie de la cataracte9).
Membrane épirétinienne (GL2026 CQ4) : vitrectomie. Une amélioration de l’acuité visuelle peut être attendue chez les patients avec une ligne EZ continue. En cas de ligne EZ discontinue, la récupération est limitée. Des cas d’atrophie maculaire sévère à long terme ont été rapportés ; une évaluation dans un centre spécialisé est souhaitable 9).
Trou maculaire : la vitrectomie est le seul traitement curatif. Les résultats postopératoires sont peu documentés 9).
Soutien et réadaptation
Soins basse vision : basse vision → loupes, loupes électroniques, tablettes ; photophobie → lunettes filtrantes ; rétrécissement du champ visuel → canne blanche ; vision de loin → monoculaire ; lunettes d’aide à la vision nocturne. Un soutien individualisé en fonction du champ visuel et de l’acuité est important. Il est recommandé d’utiliser Smart Site (présentation des services de consultation basse vision dans chaque région).
Conseil génétique : assuré par un généticien clinicien et un conseiller en génétique certifié. Les consultations portent souvent sur l’estimation du risque de récidive, les études, l’emploi, le mariage et la procréation. Il est possible de recevoir un conseil génétique sans avoir subi de diagnostic génétique.
Système des maladies rares : en tant que maladie rare désignée, le système d’aide aux frais médicaux est disponible 9). Envisagez également l’obtention d’un carnet de handicap et d’une aide médicale à l’autonomie.
Vorétigène néparvovec (Luxturna) : médicament de thérapie génique administrable aux patients présentant des variants pathogènes bialléliques du gène RPE65 et des cellules rétiniennes viables suffisantes. Approuvé au Japon en 2023 9). Dans l’essai de phase III américain (301), 31 patients ont été inscrits ; l’analyse mITT (20 dans le groupe intervention, 9 dans le groupe témoin) a montré une amélioration significative du MLMT et du FST à lumière blanche par rapport au groupe témoin 18). Dans l’essai de phase III national (A11301), une augmentation de la sensibilité FST et une expansion du champ visuel ont été confirmées chez 4 patients japonais 19). L’effet d’amélioration de l’acuité visuelle est faible, et une atrophie choroïdienne rétinienne a été rapportée chez plus de 20 % des patients comme complication à long terme 9).
Effectuer les examens suivants tous les 6 mois à 1 an : acuité visuelle, lampe à fente, fond d’œil, champ visuel Humphrey (HFA 10-2), OCT9).
QQuels médicaments sont utilisés pour traiter l'œdème maculaire ?
A
Le traitement de première intention de l’EMC-RP est un inhibiteur de l’anhydrase carbonique (IAC), administré sous forme de collyre au dorzolamide ou d’acétazolamide par voie orale 9). Une amélioration de l’épaisseur maculaire centrale est obtenue dans environ 40 % des cas, mais une récidive survient dans environ 30 % des cas. En cas d’échec des IAC, des injections intravitréennes de triamcinolone acétonide ou un implant intravitréen de dexaméthasone (Ozurdex) peuvent être envisagés. Les anti-VEGF ne sont pas recommandés dans l’EMC-RP. Il convient de noter que tous ces traitements sont utilisés hors AMM.
La voie finale commune de la mort des photorécepteurs dans la RP est l’apoptose. Bien que les types de mutations génétiques soient divers, ils convergent finalement vers une voie commune de mort cellulaire.
Dégénérescence secondaire des cônes après les bâtonnets
Dans la RP, le schéma typique est une dégénérescence et une disparition des bâtonnets en premier, suivies d’une dégénérescence secondaire des cônes 7). On pense que les cônes dépendent d’un facteur de survie produit par les bâtonnets (RdCVF : Rod-derived Cone Viability Factor) ; ainsi, après la perte des bâtonnets, les cônes perdent également leur fonction 7)11).
La rétine est l’un des tissus les plus actifs sur le plan métabolique ; elle convertit 80 à 90 % du glucose en lactate par glycolyse aérobie (effet Warburg). Les cônes sont plus vulnérables au stress métabolique que les bâtonnets, et cette vulnérabilité métabolique contribue également à la dégénérescence secondaire des cônes 11).
L’inflammation est également reconnue comme un facteur majeur de progression de la RP ; l’activation de la microglie et l’infiltration de macrophages aggravent les lésions rétiniennes 11). Le stress oxydatif agit également comme un moteur biologique de la dégénérescence secondaire des cônes.
Les mécanismes de dégénérescence diffèrent selon le gène impliqué.
Mutations RHO : La rhodopsine mal repliée induit un stress du réticulum endoplasmique → réponse aux protéines mal repliées (UPR) → apoptose11)
Mutation REEP6 : REEP6 code pour une protéine impliquée dans le maintien de la morphologie du RE. Les mutations pathogènes entraînent la formation d’inclusions intracytoplasmiques du RE dans le segment externe des bâtonnets, conduisant à la dégénérescence des photorécepteurs4)
Mutation RPGR : RPGR est impliqué dans la structure axonémale des cils primaires ; les mutations perturbent le transport des substances vers le segment externe des photorécepteurs1)
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
La thérapie génique est l’approche la plus prometteuse pour le traitement des maladies rétiniennes héréditaires8).
Luxturna (vorétigène néparvovec) : Médicament de thérapie génique administrable aux patients présentant des variants pathogènes bialléliques du gène RPE65. Dans l’essai de phase III américain (étude 301), 31 patients ont été inscrits ; l’analyse mITT (20 dans le groupe intervention, 9 dans le groupe témoin) a montré une amélioration significative du MLMT et du FST en lumière blanche par rapport au groupe témoin18). Dans l’essai de phase III national (étude A11301), une augmentation de la sensibilité FST et une expansion du champ visuel ont été confirmées chez 4 patients japonais19). Approuvé au Japon en 2023, il constitue un pont entre le traitement standard et la thérapie expérimentale.
Thérapie génique pour RPGR : La thérapie génique médiée par AAV pour la RP liée à l’X due à des mutations RPGR a progressé jusqu’aux essais cliniques de phase I/II/III8).
CRISPR/Cas9 : Des recherches sont en cours pour la correction directe de mutations pathogènes ou l’inactivation de mutations dominantes négatives8).
RdCVF (facteur de survie des cônes dérivé des bâtonnets) et thérapie de protection des cônes
Le RdCVF est une protéine sécrétée par les bâtonnets qui maintient la survie des cônes7)11). Des essais cliniques de thérapie de protection des cônes utilisant le RdCVF sont en cours, et cette approche est considérée comme une stratégie thérapeutique indépendante pour préserver la fonction des cônes après la dégénérescence des bâtonnets.
La NAC est un médicament qui supprime le stress oxydatif ; un essai de phase I a rapporté une amélioration de l’acuité visuelle17). En 2025, un essai de phase III est en cours.
Potentiel de réutilisation des glucocorticoïdes (dexaméthasone)
Des études in vivo récentes (modèle de souris rd10) ont démontré que la dexaméthasone intravitréenne protège les photorécepteurs à cône et l’épithélium pigmentaire rétinien 11). Les glucocorticoïdes ont un fort potentiel de repositionnement en tant que traitement indépendant de la mutation. Cependant, les preuves actuelles se limitent aux modèles animaux, et une validation supplémentaire est nécessaire pour une application clinique chez l’homme.
Transplantation rétinienne dérivée de cellules iPS et rétine artificielle
Transplantation rétinienne dérivée de cellules iPS : La recherche sur la transplantation de feuillets de photorécepteurs fabriqués à partir des propres cellules iPS du patient progresse.
Rétine artificielle (prothèse rétinienne) : Dispositif de stimulation électrique pour la RP terminale. L’Argus II et d’autres sont commercialisés à l’étranger, et des essais cliniques de stimulation suprachoroidienne transrétinienne sont en cours au Japon.
QLa thérapie génique est-elle disponible au Japon ?
A
Le vorétigène néparvovec (Luxturna) a été approuvé au Japon en 2023, mais uniquement pour les dystrophies rétiniennes avec variants pathogènes bialléliques du gène RPE65 9)18)19). La thérapie génique pour la RP avec d’autres mutations, y compris les mutations RPGR, est encore en phase d’essai clinique 7) et n’est pas approuvée comme traitement courant au Japon.
QComment accéder aux traitements expérimentaux ?
A
La participation aux essais cliniques est limitée aux études officielles approuvées par le comité d’éthique de l’établissement médical. En plus de consulter le médecin traitant, vous pouvez rechercher des informations sur les essais sur le site d’information sur les essais cliniques du Centre national du cancer (jRCT) ou sur clinicaltrials.gov américain.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.