L’achromatopsie (ACHM) est une maladie rétinienne héréditaire rare, bilatérale, caractérisée par la perte de fonction des trois types de photorécepteurs à cône. Elle est également appelée « monochromatisme aux bâtonnets » ou « cécité totale aux couleurs »1).
La prévalence mondiale est estimée à environ 1 personne sur 30 0001). La transmission est autosomique récessive, sans anomalies systémiques associées, et l’espérance de vie est normale.
Il existe deux formes d’ACHM : complète et incomplète. La forme complète se caractérise par une absence totale de fonction des cônes ; dans la forme incomplète, au moins un sous-type de cône conserve une fonction résiduelle, avec une acuité visuelle d’environ 20/40 à 20/120, une photophobie et un nystagmus moins sévères2).
Six gènes sont responsables (CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6), et l’identification du gène causal est possible dans plus de 90 % des cas1)2). Les gènes CNGA3 et CNGB3 représentent à eux seuls 80 à 90 % des cas.
Notez que les anomalies générales de la vision des couleurs (dyschromatopsies congénitales) sont dues à une anomalie d’un ou deux types de photopigments coniques et n’affectent que la vision des couleurs. L’ACHM diffère fondamentalement car tous les cônes sont non fonctionnels, entraînant une baisse de l’acuité visuelle, un nystagmus et une photophobie.
Il existe un cas connu d’effet fondateur sur l’île de Pingelap (Micronésie). Après un typhon dans les années 1700, la mutation CNGB3 (p.S435F) s’est répandue parmi la population décimée, atteignant une prévalence d’environ 10 % et un taux de porteurs d’environ 30 %1)2).
QQuelle est la différence entre l'achromatopsie et les anomalies générales de la vision des couleurs (dyschromatopsie) ?
A
Les dyschromatopsies congénitales générales sont dues à une anomalie d’un ou deux types de photopigments coniques, n’affectant que la vision des couleurs, et l’acuité visuelle est normale. Dans l’achromatopsie, les trois types de cônes sont non fonctionnels, ce qui entraîne, en plus de la perte de la vision des couleurs, une baisse de l’acuité visuelle (environ 0,1 ou moins), un nystagmus, une photophobie et une héméralopie, ce qui la distingue fondamentalement.
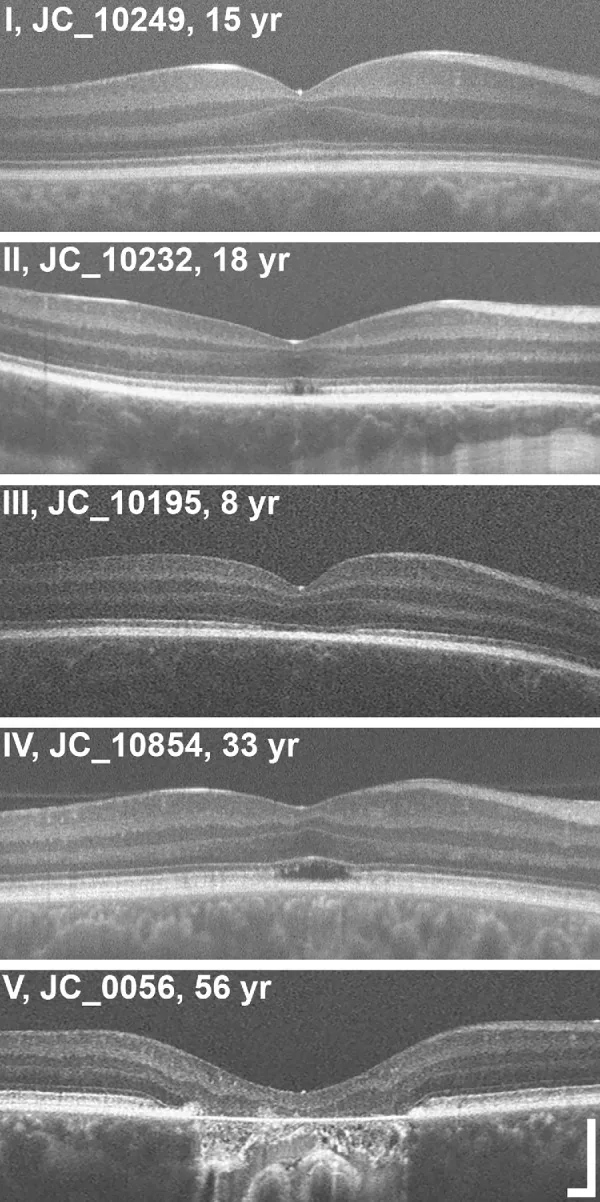
Grissim G, et al. Longitudinal Assessment of OCT-Based Measures of Foveal Cone Structure in Achromatopsia. Invest Ophthalmol Vis Sci. 2024. Figure 1. PMCID: PMC11005076. License: CC BY.
Classification en cinq grades de l’EZ par OCT : EZ normal (I), rupture de l’EZ (II), disparition de l’EZ (III), zone de faible réflectivité (IV), atrophie de la couche externe de la rétine et EPR (V). Cela correspond aux anomalies de la zone ellipsoïde traitées dans la section « 2. Principaux symptômes et signes cliniques ».
Les symptômes de l’ACHM commencent à apparaître dans les premières semaines après la naissance.
Baisse de l’acuité visuelle : dans la forme complète, elle est inférieure à 20/200 (environ 0,1) ; dans la forme incomplète, elle est d’environ 20/80 (environ 0,25).
Photophobie (sensibilité à la lumière) : 38 % des patients interrogés l’ont citée comme le symptôme le plus important2). La fonction visuelle diminue considérablement dans les environnements lumineux.
Héméralopie (cécité diurne) : la fonction visuelle diminue en lumière vive, tandis qu’elle est relativement bonne dans les environnements faiblement éclairés.
Achromatopsie/déficit de la vision des couleurs : Perte de la vision des couleurs sur les trois axes1). Les planches d’Ishihara sont presque illisibles sauf la planche de démonstration. Le test Panel D-15 montre des axes de confusion obliques circulaires.
Nystagmus pendulaire : Apparaît dans les premières semaines de vie. Tendance à s’améliorer avec l’âge et diminue lors de la vision de près.
Association d’hypermétropie : Hypermétropie fréquente, mais certaines personnes présentent une large gamme d’erreurs de réfraction incluant la myopie.
Réponse pupillaire paradoxale : Constriction pupillaire initiale dans l’obscurité, signe caractéristique. Indice important pour le diagnostic chez l’enfant.
Examen du fond d’œil : Initialement presque normal. L’angiographie à la fluorescéine montre également des résultats presque normaux. Avec le temps, des modifications et une atrophie de l’épithélium pigmentaire rétinien (EPR) peuvent survenir. On observe souvent une absence de réflexe maculaire et une dégénérescence maculaire. L’OCT montre une irrégularité et une indistinction de la zone ellipsoïde (jonction segment interne/externe des photorécepteurs) et de la zone d’interdigitation (extrémité des segments externes des cônes) dans la couche externe de la rétine.
Électrorétinographie : La réponse des cônes à l’ERG photopique est considérablement réduite ou absente, tandis que la réponse des bâtonnets à l’ERG scotopique est normale ou presque normale1)2). Dans le type GNAT2, les cônes S sont relativement mieux préservés que dans les types CNGA3/CNGB3.
Hypoplasie fovéale : Présente dans 60 à 70 % des cas de mutations CNGA3/CNGB32).
FAF (autofluorescence du fond d’œil) : Quatre motifs existent : normal, augmentation du signal central, diminution du signal central, anneau hyperfluorescent avec hypofluorescence centrale2).
Stadification OCT : Évaluation des modifications structurelles de la couche externe de la fovéa en cinq stades3).
Maintien de la couche externe de la rétine (hyperréflectivité de la ELM, aplatissement de la EZ)
Stade 2
Destruction de la zone ellipsoïde (EZ)
Stade 3
Apparition d’un espace optiquement vide
Stade 4
Espace optiquement vide + destruction partielle de l’EPR
Stade 5
Disparition de la couche nucléaire externe et/ou destruction complète de l’EPR
Dans une étude de suivi sur 10 ans, malgré une meilleure acuité visuelle corrigée stable (20/400 à 20/200), une progression structurelle a été confirmée par OCT (élargissement de l’espace optiquement vide : œil droit 246×59 μm, œil gauche 326×53 μm)3).
Des foyers hyperréflectifs apparaissent plus de 3 ans avant les modifications de l’EZ, suggérant qu’ils pourraient être un marqueur précoce de la progression de la maladie3).
AOSLO (ophtalmoscopie laser à balayage avec optique adaptative) : La mosaïque de cônes présente des espaces sombres, un espacement inter-cônes accru et une densité de cônes réduite. Il n’y a pas de différence significative entre les types CNGA3 et CNGB3, tandis que le type GNAT2 montre une préservation relative des cônes2).
Test de vision des couleurs : Le test d’Ishihara est presque illisible en dehors de la planche de démonstration. Le Panel D-15 montre un motif d’erreur sur l’axe scotopique (entre deutane et tritane). L’anomaloscope montre une pente raide et n’inclut pas la zone d’égalisation normale.
QL'acuité visuelle dans l'achromatopsie s'aggrave-t-elle avec l'âge ?
A
La meilleure acuité visuelle corrigée reste souvent stable à long terme. Cependant, l’évaluation structurelle par OCT peut montrer une progression au fil du temps (destruction de la zone ellipsoïde, élargissement de l’espace optiquement vide)3). Une dissociation entre fonction et structure est caractéristique, et un suivi régulier par examen est important.
Il s’agit d’une maladie autosomique récessive. Si les deux parents sont porteurs, le risque pour l’enfant de développer la maladie est de 25 %1). La maladie survient souvent chez des enfants de parents apparemment sains, et il n’y a souvent pas d’antécédents familiaux.
Un mode de transmission non mendélien par disomie uniparentale (UPD) paternelle a également été rapporté. Dans un cas où un homozygote pour CNGA3 c.778G>C (p.D260H) a été établi par UPD, aucune mutation n’a été détectée chez la mère 6). De tels exemples ont des implications importantes pour l’évaluation du risque de récurrence dans le conseil génétique.
Type de mutations : Principalement des mutations faux-sens. Le domaine transmembranaire S4 est un point chaud.
Distribution géographique : CNGA3 représente plus de 80 % au Moyen-Orient et en Chine.
CNGB3
Chromosome : 8q21.3
Fonction : Sous-unité bêta du canal CNG
Fréquence : Environ 50 % des cas 1)2)
Type de mutations : Principalement des mutations non-sens, de décalage du cadre de lecture et d’épissage. c.1148delC est la mutation la plus fréquente.
Distribution géographique : CNGB3 représente plus de 50 % en Europe et en Amérique.
Autres gènes
GNAT2 (1p13.3) : transducine α des cônes. Environ 2%. Forme relativement légère avec préservation de la couche des photorécepteurs2)
PDE6C (10q23.33) : sous-unité α de la PDE des cônes. Forme sévère à début précoce5)
PDE6H (12p12.3) : sous-unité γ de la PDE des cônes. Extrêmement rare1)
ATF6 (1q23.3) : facteur de transcription de la réponse au stress du réticulum endoplasmique. Environ 2%. Mécanisme n’impliquant pas directement la phototransduction1)2)
Allèle à pénétrance réduite : CNGB3 c.1208G>A (p.R403Q) conserve une fonction partielle, conduisant à un phénotype léger2).
Caractéristiques des mutations PDE6C : Dans 4 cas avec des nouvelles mutations (c.1670G>A, c.2192G>A), la triade nystagmus, photophobie et trouble de la vision des couleurs était présente chez tous. L’électrorétinogramme a montré une disparition de la réponse photopique et du flicker à 30 Hz, avec une réponse scotopique normale. Les hétérozygotes composites présentaient un phénotype plus sévère5).
Hérédité digénique : Il existe de rares cas avec des mutations à la fois dans CNGA3 et CNGB31).
QPourquoi un enfant peut-il développer la maladie même si ses parents ne sont pas achromates ?
A
L’achromatopsie étant autosomique récessive, même si les parents sont chacun porteurs d’une copie du gène muté (hétérozygotes), ils sont en apparence sains et ont une vision des couleurs normale. Chez un couple de porteurs, il y a 25% de risque que l’enfant hérite de deux copies de la mutation et développe la maladie1).
En présence de nystagmus, photophobie et baisse de l’acuité visuelle dans les premières semaines de vie, un bilan pluridisciplinaire orienté vers l’ACHM est nécessaire. Le diagnostic de certitude repose sur le test génétique.
Électrorétinogramme
Valeur diagnostique : Gold standard
Résultats de la forme complète : La réponse des cônes est absente ou fortement réduite à l’électrorétinogramme photopique. La réponse des bâtonnets à l’électrorétinogramme scotopique est normale à presque normale1)2)
Résultats de la forme incomplète : Une faible réponse des cônes correspondant à la fonction résiduelle des cônes est détectée
Caractéristiques du type GNAT2 : Les cônes S sont relativement mieux préservés que dans le type CNGA3/CNGB3
OCT
Signification diagnostique : Standard pour l’évaluation structurelle
Éléments évalués : Stadification en 5 niveaux des couches externes de la fovéa, détection de l’hypoplasie fovéolaire2)3)
Suivi : Une progression structurelle peut survenir même en l’absence de changement fonctionnel, d’où l’importance d’un suivi régulier
Examens complémentaires : Évaluation multimodale combinant FAF et AOSLO
Tests génétiques
Signification diagnostique : Indispensable pour un diagnostic de certitude
Méthode : Panel ciblé de 6 gènes (CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)2)
Taux de résolution : Identification du gène causal possible dans plus de 90% des cas1)
Participation aux essais cliniques : Un diagnostic moléculaire est obligatoire pour participer aux essais cliniques de thérapie génique
L’hypermétropie est fréquente, mais diverses anomalies de réfraction sont possibles. La correction par lunettes ou lentilles de contact est importante pour maximiser l’acuité visuelle. En cas d’amblyopie associée, envisager l’occlusion ou l’atropine.
Les verres teintés pour réduire la photophobie améliorent considérablement la qualité de vie. Selon une enquête, 96 % des patients préfèrent les filtres gris aux filtres rouges, et 74 % préfèrent les filtres gris en extérieur2). Le choix doit être adapté aux préférences et à l’environnement d’activité de chaque patient.
Une explication et un soutien par des spécialistes sont nécessaires concernant les caractéristiques de l’hérédité autosomique récessive, le risque de récidive et l’importance du diagnostic génétique. Il est également recommandé de subir un diagnostic génétique en vue de participer à de futurs essais cliniques de thérapie génique.
QQuelle couleur de verres filtrants est la plus adaptée ?
A
Dans une enquête auprès des patients, 96 % ont préféré les filtres gris aux filtres rouges, et 74 % ont préféré les filtres gris en extérieur 2). Cependant, comme il existe des différences individuelles, il est souhaitable d’essayer différents filtres et de choisir celui qui convient à son environnement d’activité en consultation avec un ophtalmologiste ou un spécialiste de la basse vision.
La cascade de transduction photique dans les cônes normaux est la suivante 1).
Obscurité: La concentration intracellulaire de cGMP est élevée, les canaux CNG sont ouverts, Na⁺ et Ca²⁺ entrent, la cellule reste dépolarisée et libère du glutamate en continu.
Exposition à la lumière: L’opsine (photopigment) est activée → la transducine (protéine G) est activée → la phosphodiestérase (PDE) est activée → le cGMP est dégradé → les canaux CNG se ferment → hyperpolarisation → inhibition de la libération de glutamate.
Rétroaction négative: La GCAP (protéine activatrice de la guanylate cyclase) se lie au Ca²⁺ et inhibe l’activité de la retGC, régulant la production de cGMP.
Mutation CNGA3: Les mutations faux-sens perturbent le repliement, le transport intracellulaire et l’intégration membranaire de la protéine 1)7). Le domaine transmembranaire S4 est un point chaud de mutation. Plus de 150 mutations faux-sens ont été rapportées, dont 103 dont la pathogénicité n’était pas établie, mais l’analyse structure-fonction 3D suggère que 86,4 % ont des conséquences fonctionnelles similaires aux mutations pathogènes connues 7).
Mutation CNGB3: Les mutations non-sens et de décalage de cadre produisent des protéines canal tronquées ou non fonctionnelles 1). En l’absence de CNGB3, les canaux homomériques CNGA3 persistent, ce qui peut préserver une fonction conique résiduelle.
Mutation ATF6: Facteur de transcription impliqué dans la réponse aux protéines mal repliées du réticulum endoplasmique (UPR), sans implication directe dans la cascade de transduction photique 1)2). Le mécanisme pathogène diffère des autres gènes et est encore à l’étude.
Le canal CNG a une structure tétramérique (CNGA3 × 3 et CNGB3 × 1, ou 2:2 selon certains rapports), chaque sous-unité possédant six domaines transmembranaires, un domaine de liaison aux nucléotides cycliques, une région de liaison C et un domaine formant le pore 1).
Mécanisme de dégénérescence des cônes et progression
Le développement et la morphologie postnatals des cônes sont presque normaux, et la dégénérescence commencerait au début de l’âge adulte. L’accumulation de cGMP est impliquée dans le processus dégénératif, et une progression plus rapide dans les zones riches en cônes S a été démontrée dans des modèles animaux 1).
Traditionnellement, l’ACHM était considérée comme une maladie non progressive. Cependant, l’observation à long terme par OCT a montré que, bien que la meilleure acuité visuelle corrigée reste presque stable, des changements structurels (rupture de la zone ellipsoïde, élargissement des espaces optiques) progressent 3). Cette dissociation fonction-structure est importante pour évaluer la fenêtre thérapeutique de la thérapie génique.
Dans la zone sans bâtonnets de la fovéa (région dense en cônes sans bâtonnets), la perte de cônes est marquée, tandis que dans la région parafovéale, les bâtonnets contribuent à la bande EZ, compensant ainsi la fonction 3).
7. Recherches récentes et perspectives futures (rapports en phase de recherche)
L’essai NCT03001310 (AAV8-hCARp.hCNGB3) portant sur 23 patients (11 adultes et 12 enfants) a montré une sécurité acceptable. Une amélioration de la vision des couleurs a été observée chez 6/23 patients, une amélioration de la photophobie chez 11/20, et une amélioration de la qualité de vie (QdV) chez 21/23. Une tendance à l’augmentation de l’inflammation intraoculaire a également été observée aux doses élevées2).
Dans l’essai AAV8.CNGA3 (RD-CURE), 9 patients ont reçu 3 doses (1×10¹⁰ à 1×10¹¹ vg/œil). Les données à 1 et 3 ans ont montré une tendance à l’amélioration de l’acuité visuelle et de la sensibilité au contraste, sans atteindre la signification statistique. La sécurité était bonne1)2).
McKyton et al. (2021) ont réalisé une cartographie visuelle corticale par IRMf après injection sous-rétinienne de AAV2tYF-PR1.7-hCNGA3 (NCT02935517) chez deux adultes atteints d’ACHM liée à CNGA34). L’œil traité a montré une tolérance à la lumière 5 fois supérieure (amélioration spectaculaire de la photophobie), une acquisition de la détection du rouge, et une réduction de la taille du champ récepteur de population (pRF) (suggérant une amélioration de la résolution spatiale). En revanche, aucune activation des zones corticales spécifiques à la couleur (comme V4) n’a été observée, et l’électrorétinogramme plein champ n’a pas détecté de réponse des cônes. Les patients ont rapporté des améliorations dans la vie quotidienne, telles qu’un « sentiment de sécurité accru aux passages piétons », « absence de besoin de loupe », et « absence de besoin de lunettes de soleil à l’extérieur ».
Plusieurs modèles animaux ont confirmé la récupération fonctionnelle après remplacement génique1)2).
Modèle murin: Récupération jusqu’à 80% de la normale à l’électrorétinogramme.
Modèle canin (CNGB3): Récupération de l’électrorétinogramme à scintillement des cônes après 2,5 ans de suivi post-remplacement génique. Amélioration comportementale dans des environnements de 25 lux ou plus.
Modèle ovin (CNGA3): Amélioration à long terme pendant au moins 6 ans après remplacement génique.
Primates non humains (PDE6C): Récupération fonctionnelle confirmée.
Concernant la fenêtre thérapeutique efficace, des études animales ont montré qu’un traitement précoce est plus efficace. Les souris âgées ont montré une réponse médiocre, mais un prétraitement au CNTF (facteur neurotrophique ciliaire) a permis une récupération fonctionnelle chez les chiens âgés1)2).
Un essai clinique (NCT04041232) du phénylbutyrate de glycérol (PBA) ciblant la réponse au stress du réticulum endoplasmique est en cours2). Cette approche vise à atténuer le mauvais repliement du réticulum endoplasmique plutôt qu’à remplacer le gène.
Actuellement, on en est au stade des essais de phase I/II de sécurité et d’efficacité. Une réduction de la photophobie et une certaine amélioration de la sensibilité à la lumière ont été rapportées, mais une restauration complète de la vision des couleurs n’a pas été atteinte2)4). Des études animales suggèrent qu’un traitement précoce pourrait être efficace, mais les données à long terme chez l’homme sont encore limitées. Il est important de suivre les progrès de la recherche et de mettre régulièrement à jour les informations avec le médecin traitant.
McKyton A, Averbukh E, Marks Ohana D, Levin N, Banin E. Cortical Visual Mapping following Ocular Gene Augmentation Therapy for Achromatopsia. J Neurosci. 2021;41(35):7363-7371. doi:10.1523/JNEUROSCI.3222-20.2021. PMID:34349002; PMCID:PMC8412991.
Madeira C, Godinho G, Grangeia A, Falcão M, Silva R, Carneiro Â, et al. Two Novel Disease-Causing Variants in the PDE6C Gene Underlying Achromatopsia. Case reports in ophthalmology. 2021;12(3):749-760. doi:10.1159/000512284. PMID:34720973; PMCID:PMC8460892.
Kohl S, Baumann B, Dassie F, et al. Paternal Uniparental Isodisomy of Chromosome 2 in a Patient with CNGA3-Associated Autosomal Recessive Achromatopsia. Int J Mol Sci. 2021;22(15):7842. doi:10.3390/ijms22157842. PMID:34360608; PMCID:PMC8346044.
Rasmussen DK, Sun YJ, Franco JA, et al. Structure-function analysis of CNGA3-associated achromatopsia patient variants complements clinical genomics in pathogenicity determination. Orphanet J Rare Dis. 2025;20:261. doi:10.1186/s13023-025-03792-3.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.