CNGA3
염색체: 2q11.2
기능: CNG 채널 알파 서브유닛
빈도: 증례의 약 25~50% 1)2)
돌연변이 패턴: 주로 미스센스 돌연변이. S4 막관통 도메인이 핫스팟입니다.
지리적 분포: 중동과 중국에서는 CNGA3가 80% 이상을 차지합니다.

전색맹(Achromatopsia; ACHM)은 세 가지 유형의 원추세포 기능이 모두 상실되는 드문 양안 유전성 망막 질환입니다. 간체 단일색형 색각(rod monochromatism) 또는 완전 색맹(total color blindness)이라고도 합니다 1).
전 세계 유병률은 약 30,000명당 1명으로 추정됩니다 1). 상염색체 열성 유전 형태를 취하며, 전신 이상을 동반하지 않고 평균 수명도 정상입니다.
ACHM에는 완전형(complete)과 불완전형(incomplete)의 두 가지 유형이 있습니다. 완전형은 모든 원추 기능이 결여되고, 불완전형은 적어도 하나의 원추 아형이 잔여 기능을 가지며 시력은 20/40~20/120 정도이고 눈부심과 안진도 경미합니다 2).
원인 유전자는 6종(CNGA3, CNGB3, GNAT2, PDE6C, PDE6H, ATF6)이며, 90% 이상의 증례에서 원인 유전자 동정이 가능합니다 1)2). CNGA3와 CNGB3 두 유전자만으로 전체의 80~90%를 차지합니다.
일반적인 색각 이상(선천성 색각 이상)은 1~2종류의 원추세포 시색소 이상으로 인해 발생하며 색각에만 영향을 미칩니다. ACHM은 모든 원추세포가 기능하지 않기 때문에 시력 저하, 안진증, 눈부심을 동반한다는 점에서 본질적으로 다릅니다.
핑겔랍 환초(미크로네시아)의 창시자 효과로 알려진 사례가 있습니다. 1700년대 태풍 이후 인구가 급감한 섬 주민들 사이에서 CNGB3 돌연변이(p.S435F)가 퍼져 유병률은 약 10%, 보인자율은 약 30%에 이릅니다1)2).
ACHM의 증상은 생후 수주 이내에 나타나기 시작합니다.
역설적 동공 반응: 암소에서 동공의 초기 수축을 보이는 특징적 소견. 소아 진단에서 중요한 단서가 됨.
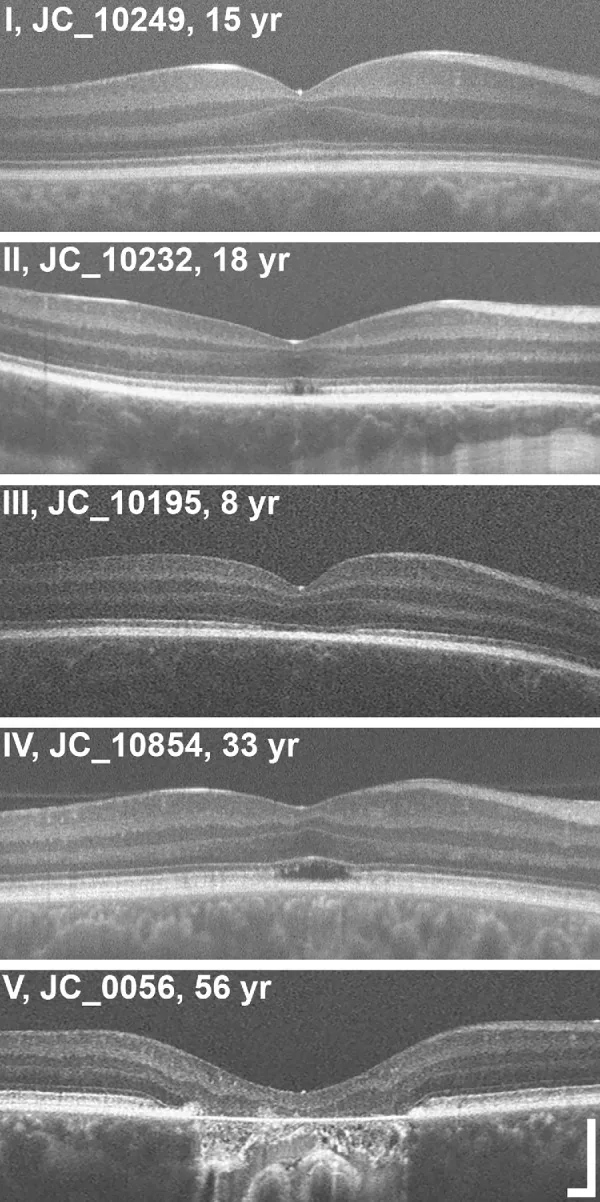
안저 소견: 초기에는 거의 정상으로 보임. 형광 안저 조영술에서도 거의 정상 소견. 경과에 따라 망막색소상피(RPE)의 반점 변화 및 위축이 발생할 수 있음. 황반 반사 소실 및 황반 변성이 자주 관찰됨. OCT에서 망막 외층의 타원체대(ellipsoid zone) 및 간상체 외절 첨단부(interdigitation zone)가 불규칙하고 불명확해짐.
망막전위도 소견: 명순응 망막전위도에서 원추 반응이 현저히 감소 또는 소실되고, 암순응 망막전위도에서 간체 반응은 정상~거의 정상을 보임1)2). GNAT2형에서는 S-원추가 CNGA3/CNGB3형보다 비교적 보존됨.
중심와 형성부전: CNGA3/CNGB3 변이에서 60~70%에서 관찰됨2).
FAF (안저 자가형광): 4가지 패턴이 존재: 정상, 중심 신호 증가, 중심 신호 감소, 과형광 고리 + 중심 저형광2).
OCT 병기: 중심와 외층의 구조적 변화를 5단계로 평가함3).
| 병기 | OCT 소견 |
|---|---|
| 1기 | 외층 망막 보존 (ELM 과반사, EZ 편평화) |
| 2기 | 타원체대(EZ) 파괴 |
| 3기 | 광학적 공간(optically empty space) 출현 |
| 4기 | 광학적 공간 + 부분적 RPE 파괴 |
| 5기 | 외핵층 소실 및/또는 완전한 RPE 파괴 |
10년 추적 연구에서 최대교정시력이 안정적(20/400~20/200)임에도 불구하고 OCT에서 구조적 진행(광학적 공간 확대: 우안 246×59 μm, 좌안 326×53 μm)이 확인되었습니다3).
고반사 초점(hyperreflective foci)이 타원체대 변화보다 3년 이상 먼저 나타나며, 질환 진행의 조기 지표가 될 가능성이 시사됩니다3).
AOSLO(적응광학 주사레이저 검안경): 원뿔세포 모자이크에 암점 영역, 원뿔세포 간격 증가, 원뿔세포 밀도 감소가 관찰됩니다. CNGA3형과 CNGB3형 사이에 유의한 차이는 없으며, GNAT2형은 비교적 원뿔세포가 보존됩니다2).
색각 검사: 이시하라 색각검사표는 시범표 외에는 거의 판독 불가능합니다. Panel D-15에서는 암순응 축(deutan과 tritan의 중간)의 오류 패턴을 보입니다. 이상색각경에서는 급격한 기울기를 보이며 정상 등색 범위를 포함하지 않습니다.
최대교정시력은 장기적으로 대체로 안정적인 경우가 많습니다. 한편, OCT를 통한 구조적 평가에서는 연령에 따른 변화(타원체대 파괴, 광학적 공간 확대)가 진행될 수 있습니다3). 기능과 구조 사이에 괴리가 발생하는 것이 특징이며, 정기적인 검사를 통한 모니터링이 중요합니다.
상염색체 열성 유전입니다. 부모가 모두 보인자인 경우 자녀의 발병 위험은 25%입니다1). 겉보기에 건강한 부모에게서 발병하는 경우가 대부분이며, 가족력이 없는 경우도 많습니다.
부계 단친 이염색체(UPD)로 인한 비멘델 유전 패턴도 보고되었습니다. UPD로 인해 CNGA3 c.778G>C (p.D260H) 동형접합체가 형성된 증례에서 어머니에게서는 돌연변이가 검출되지 않았습니다6). 이러한 예는 유전 상담에서 재발 위험 평가에 중요한 의미를 갖습니다.
CNGA3
염색체: 2q11.2
기능: CNG 채널 알파 서브유닛
빈도: 증례의 약 25~50% 1)2)
돌연변이 패턴: 주로 미스센스 돌연변이. S4 막관통 도메인이 핫스팟입니다.
지리적 분포: 중동과 중국에서는 CNGA3가 80% 이상을 차지합니다.
CNGB3
염색체: 8q21.3
기능: CNG 채널 베타 서브유닛
빈도: 증례의 약 50% 1)2)
돌연변이 패턴: 주로 넌센스, 프레임시프트, 스플라이싱 돌연변이. c.1148delC가 가장 빈번한 돌연변이입니다.
지리적 분포: 유럽과 미국에서는 CNGB3가 50% 이상을 차지합니다.
기타 유전자
GNAT2 (1p13.3): 원뿔 트랜스듀신 알파. 약 2%. 비교적 경증이며 광수용체층이 보존됨 2)
PDE6C (10q23.33): 원뿔 PDE 알파 서브유닛. 조기 발병 중증형 5)
PDE6H (12p12.3): 원뿔 PDE 감마 서브유닛. 극히 드묾 1)
ATF6 (1q23.3): 소포체 스트레스 반응 전사인자. 약 2%. 광전달에 직접 관여하지 않는 기전 1)2)
저침투성 대립유전자: CNGB3 c.1208G>A (p.R403Q)는 부분적인 기능을 유지하여 경증 표현형을 나타냄 2).
PDE6C 돌연변이의 특징: 새로운 돌연변이(c.1670G>A, c.2192G>A)를 가진 4명의 증례에서 안진, 눈부심, 색각 장애의 삼징후가 모든 예에서 관찰됨. 망막전위도에서 명순응 및 30 Hz 플리커 소실과 암순응 정상이 확인되었으며, 복합 이형접합체는 더 중증의 표현형을 보임 5).
이중유전자 유전: CNGA3와 CNGB3 모두에 돌연변이를 가진 드문 증례도 존재함 1).
전색맹은 상염색체 열성 유전이므로, 부모가 각각 하나의 돌연변이 유전자 사본을 가진 보인자(운반자)라도 외관상 건강하고 색각도 정상입니다. 보인자끼리의 조합에서는 25%의 확률로 자녀가 두 개의 돌연변이 사본을 물려받아 발병합니다 1).
생후 수주 이내에 안진, 눈부심, 시력 저하가 나타나는 경우 ACHM을 염두에 둔 다각적인 검사가 필요합니다. 확진에는 유전자 검사가 필수적입니다.
망막전위도
OCT
유전자 검사
진단적 의의: 확진에 필수
방법: 6개 유전자(CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)의 표적 패널2)
해결률: 90% 이상에서 원인 유전자 동정 가능1)
임상시험 참여: 유전자 치료 임상시험 참여에는 분자 진단이 필수
ACHM과 유사한 임상 양상을 보이는 질환과의 감별이 중요합니다2).
현재 ACHM에 대해 승인된 근치적 치료법은 없습니다1)2). 치료는 대증 요법이 중심이 됩니다.
원시가 많지만 다양한 굴절 이상을 동반하므로 안경이나 콘택트렌즈로 교정하는 것이 시력 최대화에 중요합니다. 약시가 동반된 경우 가림 요법이나 아트로핀 치료를 고려합니다.
눈부심 감소를 위한 차광 렌즈가 일상 생활의 질을 크게 개선합니다. 환자 조사에 따르면 96%가 회색 필터를 빨간색 필터보다 선호했으며, 실외에서는 74%가 회색 필터를 선호했습니다2). 개별 환자의 선호도와 활동 환경에 맞춘 선택이 중요합니다.
다음과 같은 보조기구와 기술이 활용됩니다3).
상염색체 열성 유전의 특성, 재발 위험, 유전자 진단의 중요성에 대해 전문가의 설명과 지원이 필요합니다. 향후 유전자 치료 임상시험 참여를 대비하여 유전자 진단을 받는 것도 권장됩니다.
환자 조사에서 96%가 적색 필터보다 회색 필터를 선호했으며, 실외에서는 74%가 회색 필터를 선호했습니다2). 그러나 개인차가 있으므로 실제로 다양한 필터를 시도한 후, 자신의 활동 환경에 맞는 필터를 안과 의사나 저시력 전문가와 상담하여 선택하는 것이 바람직합니다.
정상 원추세포에서의 광전달 캐스케이드는 다음과 같습니다1).
CNGA3 돌연변이: 미스센스 돌연변이가 단백질 접힘, 세포 내 수송 및 막 통합을 손상시킵니다1)7). S4 막관통 도메인이 돌연변이 핫스팟입니다. 150종 이상의 미스센스 돌연변이가 보고되었으며, 103개는 병원성이 확정되지 않았으나 3차원 구조-기능 분석에 따르면 86.4%가 알려진 병원성 돌연변이와 유사한 기능적 결과를 초래하는 것으로 시사됩니다7).
CNGB3 돌연변이: 무의미 및 프레임시프트 돌연변이가 절단형 또는 기능 상실형 채널 단백질을 생성합니다1). CNGB3 결핍 시에도 잔여 CNGA3 동종채널이 존재하여 약간의 원추 기능이 유지될 가능성이 있습니다.
ATF6 돌연변이: ATF6는 소포체의 미접힘 단백질 반응(UPR)에 관여하는 전사인자이며, 광전달 캐스케이드에 직접 관여하지 않습니다1)2). 병태 기전은 다른 유전자와 다르며 현재도 해명이 진행 중입니다.
CNG 채널은 사량체 구조(CNGA3 × 3과 CNGB3 × 1, 일부 보고에서는 2:2)를 가지며, 각 서브유닛은 6개의 막관통 도메인, 고리형 뉴클레오티드 결합 도메인, C-linker 영역 및 세공 형성 도메인을 가지고 있습니다1).
출생 후 원추의 주요 발달과 형태는 정상에 가깝고, 변성은 젊은 성인기부터 시작되는 것으로 생각됩니다. cGMP 축적이 변성 과정에 관여하며, 동물 모델에서 S-원추 풍부 영역에서 더 빠른 진행이 나타납니다1).
전통적으로 ACHM은 비진행성 질환으로 간주되었습니다. 그러나 OCT 장기 관찰을 통해 최대교정시력이 거의 안정적임에도 불구하고 구조적 변화(EZ 파괴, 광학적 공간 확대)가 진행됨이 입증되었습니다3). 이러한 기능-구조 괴리는 유전자 치료의 치료 창을 평가하는 데 중요한 의미를 갖습니다.
중심와의 무간상체 영역(간상체가 없는 원뿔세포 밀집 영역)에서는 원뿔세포 소실이 현저한 반면, 부중심와에서는 간상체가 EZ 밴드에 기여하여 기능을 보상합니다3).
2024년 기준 진행 중이거나 완료된 주요 유전자 치료 임상 시험은 아래와 같습니다1)2).
| NCT 번호 | 표적 유전자 | 벡터 | 상태 |
|---|---|---|---|
| NCT03001310 | CNGB3 | AAV8-hCARp.hCNGB3 | 완료 |
| NCT02610582 | CNGA3 | AAV8-hG1.7-hCNGA3 | 완료 |
| NCT03758404 | CNGA3 | rAAV8.hCNGA3 | 모집 중 |
| NCT02599922 | CNGB3 | AAV2tYF-PR1.7-hCNGB3 (AGTC-401) | 진행 중 (모집 완료) |
| NCT02935517 | CNGA3 | AAV2tYF-PR1.7-hCNGA3 (AGTC-402) | 진행 중 (모집 완료) |
NCT03001310(AAV8-hCARp.hCNGB3)의 성인 11명, 소아 12명 총 23명을 대상으로 한 시험에서 안전성은 허용 범위 내에 있었다. 색각 개선이 6/23명, 눈부심 개선이 11/20명, 삶의 질(QoL) 개선이 21/23명에서 확인되었다. 고용량에서는 안내 염증 증가 경향도 관찰되었다2).
AAV8.CNGA3(RD-CURE) 시험에서는 9명에게 3가지 용량(1×10¹⁰~1×10¹¹ vg/안)이 투여되었다. 1년 및 3년 데이터에서 시력 및 대비 감도 개선 경향이 인정되었으나 통계적 유의성에는 도달하지 못했다. 안전성은 양호하였다1)2).
McKyton 등(2021)은 CNGA3-ACHM 성인 2명에게 AAV2tYF-PR1.7-hCNGA3(NCT02935517)를 망막하 주사한 후 fMRI를 통한 피질 시각 매핑을 수행하였다4). 치료안은 비치료안에 비해 5배의 광내성(눈부심의 극적인 개선)이 인정되었고, 적색 검출 능력 획득, population receptive field(pRF) 크기 감소(공간 해상도 향상 시사)가 확인되었다. 한편, 색각 특이적 피질 영역(V4 등)의 활성화는 인정되지 않았으며, 전시야 망막전위도에서도 원추 반응은 검출 불가능한 상태로 남아 있었다. 환자의 자가 보고에서는 “횡단보도에서 안전감 향상”, “확대경 불필요”, “야외에서 선글라스 불필요”와 같은 일상 생활 개선이 보고되었다.
여러 동물 모델에서 유전자 보충 후 기능 회복이 확인되었다1)2).
치료의 유효 시간 창에 대해서는, 젊은 나이에 치료하는 것이 더 효과적이라는 것이 동물 실험에서 나타났다. 노령 마우스에서는 반응이 좋지 않았지만, CNTF(섬모체 신경영양인자) 전처리로 노령 개에서도 기능 회복이 가능하였다1)2).
소포체 스트레스 반응을 표적으로 하는 페닐부티르산 글리세롤(PBA)의 임상 시험(NCT04041232)이 진행 중이다2). 유전자 보충이 아닌 소포체의 접힘 이상을 완화하는 접근법으로 주목받고 있다.
유전자 치료의 주요 과제는 다음과 같다1)2)4).
현재는 제I/II상 안전성·유효성 시험 단계입니다. 눈부심 감소 및 일부 광감수성 개선이 보고되었지만, 완전한 색각 회복은 달성되지 않았습니다2)4). 동물 실험에서 젊은 나이에 치료가 효과적일 가능성이 제시되었지만, 인간에서의 장기 데이터는 아직 제한적입니다. 연구 진행 상황을 주시하고 담당 의사와 정기적으로 정보를 업데이트하는 것이 중요합니다.