CNGA3
染色体: 2q11.2
功能: CNG通道α亚基
频率: 约占病例的25–50% 1)2)
突变模式: 以错义突变为主。S4跨膜结构域是热点区域。
地理分布: 在中东和中国,CNGA3占80%以上。

全色盲(ACHM)是一种罕见的双侧遗传性视网膜疾病,所有三种视锥细胞功能丧失。也称为杆体单色视或完全色盲 1)。
全球患病率估计约为每30,000人中有1人 1)。为常染色体隐性遗传,不伴全身异常,预期寿命正常。
ACHM分为完全型和不完全型两种。完全型所有视锥功能缺失;不完全型至少一种视锥亚型保留部分功能,视力约为20/40至20/120,畏光和眼球震颤较轻 2)。
已发现6种致病基因(CNGA3、CNGB3、GNAT2、PDE6C、PDE6H、ATF6),90%以上的病例可确定致病基因 1)2)。仅CNGA3和CNGB3就占所有病例的80-90%。
需要注意的是,一般的色觉异常(先天性色觉异常)是由1~2种锥体视蛋白异常引起的,仅影响色觉。ACHM则因所有锥体细胞功能丧失,本质上不同,伴有视力下降、眼球震颤和畏光。
有一个著名的案例是平格拉普环礁(密克罗尼西亚)的奠基者效应。在18世纪台风导致人口锐减后,CNGB3突变(p.S435F)在岛民中传播,患病率约10%,携带率约30%1)2)。
ACHM的症状在出生后数周内开始出现。
反常瞳孔反应: 在暗处出现瞳孔初始收缩的特征性表现。是儿童诊断的重要线索。
眼底所见: 初期几乎正常。荧光眼底造影也几乎正常。随着病程进展,可能出现视网膜色素上皮(RPE)的斑片状变化和萎缩。常可见黄斑反射消失和黄斑变性。OCT显示视网膜外层椭圆体带和嵌合体带不规则且模糊。
视网膜电图所见: 明适应视网膜电图显示锥体反应显著降低或消失,暗适应视网膜电图显示杆体反应正常或接近正常1)2)。GNAT2型中S锥体比CNGA3/CNGB3型相对保留。
中心凹发育不全: 在CNGA3/CNGB3突变中60-70%可见2)。
FAF(眼底自发荧光): 存在四种模式:正常、中心信号增强、中心信号减弱、高荧光环伴中心低荧光2)。
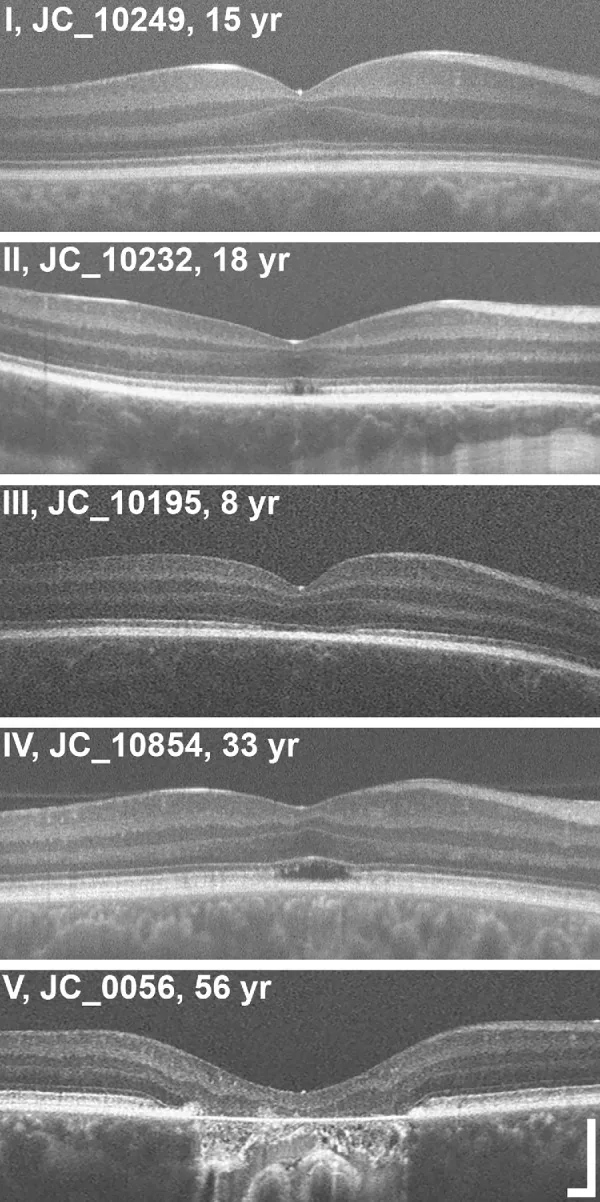
OCT分期: 中心凹外层结构变化分为5个阶段进行评估3)。
| 分期 | OCT所见 |
|---|---|
| 第1期 | 外层视网膜保留(ELM高反射、EZ扁平化) |
| 第2期 | 椭圆体带(EZ)破坏 |
| 第3期 | 出现光学空腔(optically empty space) |
| 第4期 | 光学空腔 + 部分RPE破坏 |
| 第5期 | 外核层消失和/或完全RPE破坏 |
在一项为期10年的随访研究中,尽管最佳矫正视力稳定(20/400至20/200),但OCT显示结构性进展(光学空腔扩大:右眼246×59 μm,左眼326×53 μm)3)。
高反射灶在椭圆体带变化前3年以上出现,提示其可能是疾病进展的早期标志物3)。
AOSLO(自适应光学扫描激光检眼镜):视锥细胞镶嵌中出现暗区、视锥细胞间距增大、密度降低。CNGA3型和CNGB3型之间无显著差异;GNAT2型视锥细胞相对保留2)。
色觉检查:石原色盲检查表除演示表外几乎无法辨认。Panel D-15显示暗视轴错误模式(介于红色盲和蓝色盲之间)。异常色觉镜检查显示陡峭斜率,不包含正常匹配范围。
最佳矫正视力通常长期稳定。然而,OCT的结构性评估可能显示随时间进展的变化(椭圆体带破坏、光学空腔扩大)3)。功能与结构之间出现分离是其特点,定期监测很重要。
常染色体隐性遗传。如果父母双方均为携带者,子女发病风险为25%1)。患者通常外表健康的父母所生,家族史可能缺失。
也有报道因父源单亲二倍体(UPD)导致的非孟德尔遗传模式。在一例因UPD导致CNGA3 c.778G>C (p.D260H)纯合子的病例中,母亲未检测到突变6)。此类例子对遗传咨询中的复发风险评估具有重要意义。
CNGA3
染色体: 2q11.2
功能: CNG通道α亚基
频率: 约占病例的25–50% 1)2)
突变模式: 以错义突变为主。S4跨膜结构域是热点区域。
地理分布: 在中东和中国,CNGA3占80%以上。
CNGB3
染色体: 8q21.3
功能: CNG通道β亚基
频率: 约占病例的50% 1)2)
突变模式: 以无义、移码和剪接突变为主。c.1148delC是最常见的突变。
地理分布: 在欧洲和美国,CNGB3占50%以上。
其他基因
GNAT2(1p13.3):锥体转导素α亚基。约占2%。病情相对较轻,光感受器层保留2)
PDE6C(10q23.33):锥体PDE α亚基。早发型重症5)
PDE6H(12p12.3):锥体PDE γ亚基。极为罕见1)
ATF6(1q23.3):内质网应激反应转录因子。约占2%。机制不直接参与光传导1)2)
低外显率等位基因:CNGB3 c.1208G>A (p.R403Q)保留部分功能,导致轻度表型2)。
PDE6C突变特征:在携带新突变(c.1670G>A、c.2192G>A)的4例患者中,所有病例均出现眼球震颤、畏光和色觉障碍三联征。视网膜电图显示明视和30 Hz闪烁反应消失,暗视反应正常,复合杂合子表现出更严重的表型5)。
双基因遗传:存在罕见的CNGA3和CNGB3均携带突变的病例1)。
由于全色盲是常染色体隐性遗传,父母可能各携带一个突变基因拷贝(携带者),外表健康且色觉正常。当两个携带者生育时,孩子有25%的概率继承两个突变拷贝而发病1)。
如果在出生后数周内出现眼球震颤、畏光和视力下降,需要进行以ACHM为重点的多方面检查。确诊必须依靠基因检测。
视网膜电图
OCT
基因检测
诊断意义: 确诊所必需
方法: 6个基因(CNGA3/CNGB3/GNAT2/PDE6C/PDE6H/ATF6)的目标面板2)
解决率: 超过90%的病例可确定致病基因1)
临床试验参与: 参加基因治疗临床试验必须进行分子诊断
与具有类似临床表现的疾病进行鉴别很重要2)。
目前,ACHM尚无批准的根治性治疗方法1)2)。治疗以对症治疗为主。
远视常见,但伴有广泛的屈光不正。通过眼镜或隐形眼镜矫正对最大化视力很重要。如果合并弱视,可考虑遮盖疗法或阿托品治疗。
用于减轻畏光的遮光镜片可显著改善生活质量。患者调查显示,96%的人更喜欢灰色滤光片而不是红色滤光片,74%的人在户外更喜欢灰色滤光片2)。应根据患者个人偏好和活动环境进行选择。
使用以下辅助器具和技术3)。
需要专家就常染色体隐性遗传的特点、复发风险以及基因诊断的意义进行说明和支持。也建议接受基因诊断,为将来参加基因治疗临床试验做准备。
患者调查显示,96%的人偏好灰色滤光片而非红色滤光片,户外74%的人偏好灰色滤光片2)。但由于个体差异,建议实际尝试各种滤光片后,与眼科医生或低视力专家协商选择适合自己活动环境的滤光片。
正常锥体感光细胞中的光传导级联如下1)。
CNGA3突变: 错义突变损害蛋白质折叠、细胞内运输和膜整合1)7)。S4跨膜结构域是突变热点。已报道150多种错义突变,其中103种致病性未确定,但三维结构-功能分析表明86.4%与已知致病突变具有相似的功能后果7)。
CNGB3突变: 无义和移码突变产生截短或功能丧失的通道蛋白1)。CNGB3缺失时,残留的CNGA3同源通道可能保留部分锥体功能。
ATF6突变: ATF6是参与内质网未折叠蛋白反应(UPR)的转录因子,不直接参与光传导级联1)2)。其致病机制与其他基因不同,目前仍在研究中。
CNG通道为四聚体结构(CNGA3 × 3和CNGB3 × 1,部分报道为2:2),每个亚基包含6个跨膜结构域、环核苷酸结合结构域、C-linker区和孔道形成结构域1)。
出生后锥体的主要发育和形态接近正常,变性被认为始于青年期。cGMP积累参与变性过程,动物模型显示S锥体丰富区域进展更快1)。
传统上,ACHM被认为是一种非进展性疾病。然而,OCT长期观察表明,尽管最佳矫正视力基本稳定,但结构性变化(EZ破坏、光学空隙扩大)仍在进展3)。这种功能-结构分离对评估基因治疗的治疗窗口具有重要意义。
在中心凹的无杆体区(无杆体的锥体密集区域),锥体丧失显著,而在旁中心凹,杆体通过贡献于EZ带而代偿功能3)。
截至2024年正在进行或已完成的主要基因治疗临床试验如下所示1)2)。
| NCT编号 | 靶基因 | 载体 | 状态 |
|---|---|---|---|
| NCT03001310 | CNGB3 | AAV8-hCARp.hCNGB3 | 已完成 |
| NCT02610582 | CNGA3 | AAV8-hG1.7-hCNGA3 | 已完成 |
| NCT03758404 | CNGA3 | rAAV8.hCNGA3 | 招募中 |
| NCT02599922 | CNGB3 | AAV2tYF-PR1.7-hCNGB3 (AGTC-401) | 进行中(尚未招募) |
| NCT02935517 | CNGA3 | AAV2tYF-PR1.7-hCNGA3 (AGTC-402) | 进行中(尚未招募) |
NCT03001310(AAV8-hCARp.hCNGB3)试验共纳入23名受试者(11名成人,12名儿童),安全性在可接受范围内。6/23名受试者色觉改善,11/20名畏光改善,21/23名生活质量(QoL)改善。高剂量组观察到眼内炎症增加的趋势2)。
AAV8.CNGA3(RD-CURE)试验中,9名受试者接受了三种剂量(1×10¹⁰至1×10¹¹ vg/眼)。1年和3年数据显示视力和对比敏感度有改善趋势,但未达到统计学显著性。安全性良好1)2)。
McKyton等人(2021)对两名CNGA3-ACHM成人患者进行AAV2tYF-PR1.7-hCNGA3(NCT02935517)视网膜下注射后,通过fMRI进行皮质视觉映射4)。治疗眼与非治疗眼相比,光耐受性提高5倍(畏光显著改善),获得红色检测能力,群体感受野(pRF)大小减小(提示空间分辨率提高)。然而,未观察到颜色特异性皮质区域(如V4)的激活,全视野视网膜电图仍检测不到锥体反应。患者自我报告日常生活改善,如“过马路安全感提高”、“无需放大镜”、“户外无需戴太阳镜”。
多个动物模型已证实基因补充后的功能恢复1)2)。
关于治疗的有效时间窗,动物实验表明年轻时期治疗更有效。老年小鼠反应不佳,但CNTF(睫状神经营养因子)预处理可使老年犬也能恢复功能1)2)。
针对内质网应激反应的苯丁酸甘油酯(PBA)临床试验(NCT04041232)正在进行中2)。作为一种缓解内质网折叠异常而非基因补充的方法,备受关注。
基因治疗的主要挑战如下1)2)4)。
目前处于I/II期安全性和有效性试验阶段。已有报告显示畏光减轻和部分光敏感性改善,但尚未实现完全色觉恢复2)4)。动物实验表明,年轻时治疗可能有效,但人类长期数据仍然有限。重要的是关注研究进展,并与主治医生定期更新信息。