Retinite pigmentosa (RP) é um termo abrangente para um grupo de doenças hereditárias caracterizadas por degeneração progressiva extensa dos fotorreceptores (bastonetes e cones) e do epitélio pigmentar da retina (EPR). A degeneração dos bastonetes precede a degeneração dos cones, e esse tipo é chamado de distrofia de bastonetes-cones, entendido como sinônimo de RP. Não é uma doença única, mas um grupo de doenças envolvendo mais de 100 genes.
A prevalência é de 1 a cada 4.000–8.000 pessoas, com um total de pacientes no Japão de pelo menos 30.000 (20.687 beneficiários de doença rara especificada em 2023) 9). Em relação à deficiência visual, a RP ocupa o segundo lugar (13,0%, após glaucoma com 40,7% em 2019) como causa de deficiência visual entre novos titulares de carteira de deficiência física com idade ≥18 anos, e é a principal causa de cegueira congênita 9). Foi designada como doença rara especificada sob a Lei de Doenças Raras do Japão (a partir de 1º de janeiro de 2015) 9), sendo elegível para subsídio de despesas médicas.
A RP também pode ser sindrômica, acompanhada de outras doenças sistêmicas, e é classificada sob o conceito mais amplo de ciliopatia (ciliopathy) da seguinte forma 9)2).
Ciliopatia (ciliopathy):
Síndrome de Usher (tipo 1/2/3): RP + perda auditiva; doença rara específica (AR). No tipo 1, há perda auditiva severa e disfunção vestibular desde a infância.
Mucopolissacaridose (Hurler, Hunter): Acompanhada de opacidade de fundo.
Doença de Refsum (tipo adulto e infantil):Doença peroxissomal; ataxia cerebelar, neuropatia múltipla (AR).
Síndrome de Bassen-Kornzweig: Distúrbio do metabolismo lipídico.
Doenças mitocondriais:
Síndrome de Kearns-Sayre: Oftalmoplegia externa progressiva bilateral, ptose, distúrbio de condução cardíaca.
Distrofia muscular:
Distrofia miotônica: Pode estar associada à RP
Além disso, é importante diferenciar de várias síndromes como PHARC (polineuropatia, surdez, ataxia, RP, catarata), PCARP e síndrome de Oliver-McFarlane 3).
QA retinite pigmentosa é hereditária?
A
A RP é uma doença hereditária, mas nem sempre é transmitida a todos. O risco de transmissão para os filhos varia de acordo com o padrão de herança. No tipo AD, há 50% de chance de o filho herdar a doença, enquanto nos tipos AR ou XL, o risco varia conforme o padrão. Nos casos esporádicos (48-63% do total), o risco de transmissão para a próxima geração é frequentemente relativamente baixo 9). Recomenda-se o uso de aconselhamento genético.
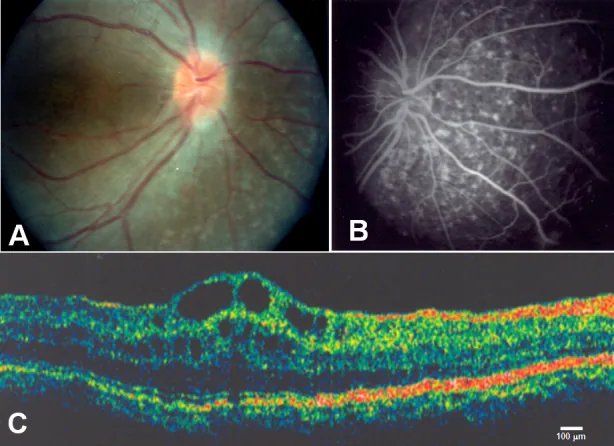
Zenteno JC, et al. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009. Figure 1. PMCID: PMC2742641. License: CC BY.
A mostra drusas do disco óptico e atrofia extensa do epitélio pigmentar da retina, B mostra fluorescência coroidal correspondente à atrofia do epitélio pigmentar da retina, C mostra edema macular cistóide e separação das camadas internas da retina na fóvea. Corresponde ao edema macular cistóide discutido na seção “2. Principais sintomas e achados clínicos”.
Os sintomas da RP mudam de acordo com o estágio de progressão. Como os bastonetes degeneram primeiro, a cegueira noturna aparece como o sintoma mais precoce.
Cegueira noturna: Diminuição da visão ou dificuldade para enxergar em locais escuros. Aparece muito precocemente porque os bastonetes degeneram primeiro 9); é percebida entre os 10 e 20 anos como dificuldade para enxergar em ambientes com pouca luz. No início, a função visual diurna é frequentemente normal.
Estreitamento do campo visual: O campo visual estreita-se gradualmente da periferia para o centro. Progride de escotoma anular para estreitamento concêntrico do campo visual (visão tubular) 9)
Diminuição da acuidade visual: Quando a degeneração dos cones progride após a dos bastonetes, a acuidade visual central também diminui. Na presença de CME, pode ocorrer diminuição moderada da acuidade visual em estágio relativamente precoce. Em alguns casos, a acuidade visual central pode ser mantida até estágios avançados.
Fotofobia (cegueira diurna): Sensibilidade excessiva à luz. É uma manifestação de disfunção dos cones. Aumenta com a progressão da degeneração dos cones. É importante diferenciar da dispersão de luz causada por catarata.
Fotopsia: Pode ocorrer devido à degeneração e perda de fotorreceptores.
Alucinações visuais (Síndrome de Charles Bonnet): Fenômeno em que pacientes com perda progressiva da visão veem paisagens ou pessoas que não existem. Não é uma experiência patológica, mas sim um fenômeno decorrente da hiperatividade do córtex visual9)
Abaixo está um guia da progressão dos sintomas de acordo com o estágio da doença.
Estágio
Sintomas principais
Faixa etária aproximada
Inicial
Cegueira noturna
10–20 anos
Intermediário
Estreitamento do campo visual (escotoma anular → centrípeto)
30–40 anos
Avançado
Diminuição da acuidade visual, anomalias de visão de cores, fotofobia
Pigmentação em espículas ósseas: Pigmentação característica que aparece da região médio-periférica à periférica (padrão de espícula óssea)
Estreitamento das artérias retinianas: Ocorre secundariamente à degeneração dos fotorreceptores
Palidez cerosa do disco óptico: Reflete a degeneração do nervo óptico
Classificação do tipo de doença divide-se em típica e atípica 9).
RP típica (distrofia de bastonetes-cones): Os bastonetes são afetados primeiro, e os cones são afetados posteriormente
Distrofia de bastonetes (subtipo): Os cones não são afetados até estágios finais; a acuidade visual central é mantida mesmo com constrição severa do campo visual
RP atípica9):
RP sem pigmento: Nenhuma pigmentação é observada
RP unilateral: Observada em apenas um olho, ou com grande diferença entre os olhos
RP setorial: Limitada a 1-2 quadrantes da retina; progressão lenta, prognóstico favorável
RP central e paracentral: Lesões retinianas e anormalidades do campo visual começam no centro
Retinopatia punctata alba: Lesões puntiformes brancas a amareladas na retina
Em crianças, os achados típicos frequentemente não estão completos, e o ERG é a chave para o diagnóstico.
Catarata subcapsular posterior (CSP): ocorre em cerca de 50% dos casos. Caracteriza-se por diminuição da acuidade visual em ambientes claros. Largura da zona elipsoide (EZ) ≥600 μm prediz boa acuidade visual pós-cirurgia de catarata (AUC 0,97)5)
Edema macular cistóide (EMC): ocorre em 10-50% dos casos, sendo a principal causa de diminuição da acuidade visual central9)
Glaucoma de ângulo fechado: crises relatadas em cerca de 1% dos casos; subluxação do cristalino devido à fragilidade das zônulas de Zinn também pode ocorrer9)
Buraco macular e esquize foveal: relativamente raros, mas podem ser indicação de vitrectomia9)
QA acuidade visual melhora após a cirurgia de catarata?
A
Na cirurgia de catarata para catarata subcapsular posterior associada à RP, se a largura da zona elipsoide (EZ) na OCT pré-operatória for ≥600 μm, pode-se esperar boa acuidade visual pós-operatória (AUC 0,97)5). A largura da EZ é um biomarcador útil para prever a função visual pré-operatória. No entanto, as zônulas de Zinn são frequentemente frágeis, exigindo atenção para contração capsular anterior e deslocamento da LIO. Como profilaxia do EMC pós-operatório (10-14%), recomenda-se o uso de colírios de esteroides e AINEs por período mais prolongado que o habitual9).
A RP é um grupo de doenças geneticamente heterogêneas causadas por mutações em mais de 100 genes9). Abaixo estão os principais genes causadores na população japonesa, de acordo com o padrão de herança.
Abaixo está uma comparação dos principais genes causadores.
Gene
Padrão de herança
Frequência/características em japoneses
EYS
AR
30-50% dos casos com gene identificado (mais frequente no tipo AR)12)13)
USH2A
AR
Segundo mais comum no tipo AR (4-9%); gene principal da síndrome de Usher12)
RHO
AD
Mais comum no tipo AD 6)
RPGR
Ligado ao X
Cerca de 70-75% no tipo XL 6)
REEP6
AR
Um dos genes causadores do tipo AR 4)
Abaixo estão detalhes adicionais sobre as características de cada gene.
EYS (Eyes Shut Homolog): Gene mais frequente em RP tipo AR em japoneses (30-50% dos casos com gene identificado) 12)13). Não é tão frequente no Ocidente, refletindo a herança genética única dos japoneses.
USH2A: Gene principal da síndrome de Usher (RP + perda auditiva), e segundo gene mais comum em RP tipo AR em japoneses após EYS (4-9%) 12)
RHO (Rodopsina): Gene causador mais comum de RP do tipo AD 6). Codifica a proteína fotorreceptora nos bastonetes.
RPGR (Regulador de GTPase da Retinite Pigmentosa): Gene causador principal de RP do tipo XL 6). Pacientes do sexo masculino com mutações no RPGR podem apresentar disfunção ciliar primária (PCD) 1).
REEP6 (Proteína 6 de Aumento da Expressão de Receptores): Um dos genes causadores de RP do tipo AR 4).
A taxa de detecção de genes causadores em testes genéticos varia conforme o padrão de herança. Para o tipo AD, 35-60%; para o tipo AR/casos esporádicos, 30-50%; e para o tipo XL, 16-36% 6).
Além disso, na RP sindrômica, que inclui síndrome de Joubert, síndrome de Bardet-Biedl, etc., mutações em genes relacionados aos cílios são frequentes, podendo haver complicações sistêmicas (doença renal, polidactilia, obesidade, etc.) 2). É importante também o diagnóstico diferencial com várias síndromes, como ataxia de Friedreich (PHARC), PCARP, síndrome de Oliver-McFarlane, entre outras 3).
QDevo fazer o teste genético?
A
O diagnóstico genético é importante para o diagnóstico definitivo, aconselhamento genético e determinação da elegibilidade para terapia gênica. O sistema de painel PrismGuide IRD (análise abrangente das sequências de éxons de 82 genes causadores de doenças hereditárias da retina) foi aprovado para cobertura de seguro em 2023, mas, a partir de junho de 2025, é destinado apenas a pacientes jovens com suspeita de IRD relacionada ao RPE65 9). Recomenda-se sua realização em conjunto com aconselhamento genético. O aconselhamento genético pode ser recebido sem a realização do diagnóstico genético.
O diagnóstico de RP é feito combinando achados clínicos, exames eletrofisiológicos, exames de imagem e testes genéticos.
Critérios diagnósticos (critérios de certificação)9) incluem os seguintes elementos:
A. Sintomas (um ou mais):
Sintomas subjetivos progressivos
Um ou mais dos seguintes: cegueira noturna, constrição do campo visual, diminuição da acuidade visual, fotofobia
B. Achados de exame (dois ou mais):
(1) Achados de fundo de olho: estreitamento dos vasos retinianos, textura retiniana grosseira, depósitos pigmentares em forma de espícula óssea, múltiplos pontos brancos, atrofia do nervo óptico, degeneração macular
Anormalidade azul-amarela adquirida é frequente; teste Panel D-15 e 100 Hue9)
Teste de adaptação ao escuro
Avaliação da função dos bastonetes
Ponto de inflexão de Kohlrausch não é detectado10)
Teste genético NGS
Diagnóstico genético
Painel PrismGuide IRD (82 genes) 9)
Os detalhes de cada exame são mostrados abaixo.
Eletrorretinografia (ERG): Essencial para o diagnóstico definitivo 6)9). A resposta dos bastonetes (ERG escotópico) diminui desde o início e, com a progressão, a resposta dos cones (ERG fotópico) também diminui. O ERG de campo total é o padrão. Frequentemente já não é registrável na primeira consulta.
OCT (Tomografia de Coerência Óptica): Avalia a largura e o padrão de desaparecimento da EZ (zona elipsoide). A largura da EZ é útil como biomarcador quantitativo da função visual e prognóstico, e também é usada para determinar a adequação da cirurgia de catarata5). O afinamento da camada nuclear externa e o desaparecimento da EZ são observados desde o início.
Autofluorescência de Fundo (FAF): Um anel hiperautofluorescente anormal (anel AF) aparece ao redor da mácula, indicando progressão da doença e retina com função residual 6).
Exame de Campo Visual: O exame de campo visual dinâmico com perímetro de Goldmann é o padrão. Com a progressão, segue-se escotoma anular → estreitamento concêntrico do campo visual 10). O programa HFA 10-2 é útil para avaliar a função dos cones centrais residuais 9).
Exame de Visão de Cores: Anormalidade adquirida azul-amarelo é frequentemente observada. Avaliada com o teste Panel D-15 e teste 100 Hue 9).
Exame de Adaptação ao Escuro: O ponto de inflexão de Kohlrausch (ponto de transição entre bastonetes e cones) não é detectado 10).
Sequenciador de Próxima Geração (NGS): O sistema de painel PrismGuide IRD pode analisar de forma abrangente as sequências de éxons de 82 genes causadores de doenças 9). Também é essencial para determinar a adequação da terapia gênica.
Atualmente, não existe tratamento curativo para RP 6)9). O tratamento visa preservar a função visual, tratar complicações e apoiar a vida social.
Proteção dos Fotorreceptores
Vitamina A (15.000 UI/dia): Relatos indicam que a administração oral retarda a deterioração do ERG em alguns por cento 14). Não melhora a acuidade visual ou o campo visual. Monitoramento da função hepática é necessário em uso prolongado. Contraindicada na gravidez devido ao risco teratogênico. Em mutações ABCA4, pode acelerar a progressão 14). Nota: a vitamina E pode acelerar a progressão, portanto, requer cautela 14).
Colírio de Unoprostona: Mostrou melhora na sensibilidade dependente da dose, mas não atingiu significância no desfecho primário do estudo de fase 2 (sensibilidade retiniana central de 2 graus) 16).
Nilvadipina (bloqueador de cálcio): Relatos de longo prazo sugerem desaceleração da progressão do defeito de campo visual15). Esses relatos são de um único centro com pequeno número de pacientes, e não houve confirmação em estudos multicêntricos.
N-acetilcisteína (NAC): Suprime o estresse oxidativo. Um estudo de fase I relatou melhora na acuidade visual17), e um estudo de fase III está em andamento em 2025.
DHA e Luteína: Protegem os fotorreceptores da mácula do estresse oxidativo. A adição de DHA à vitamina A não mostrou benefício adicional.
Helenieno (Adaptinol): Aprovado para melhora temporária do campo visual e adaptação ao escuro na RP. A avaliação de eficácia pelos padrões da medicina moderna não foi realizada.
Óculos de proteção contra luz: Reduzem o estresse oxidativo causado por UV e luz intensa. Recomenda-se uso diário.
Tratamento de Complicações
Tratamento do Edema Macular Cistóide (EMC):
A primeira linha são os inibidores da anidrase carbônica (IAC).
Use colírio de dorzolamida (Trusopt) ou acetazolamida oral (Diamox). A melhora da espessura macular central (CMT) é alcançada em cerca de 40%. A recorrência ocorre em cerca de 30% 9).
Os anti-VEGF não são recomendados no EMC associado à RP (RP-EMC) porque a produção de VEGF está reduzida 9).
Observe que nenhum deles é aprovado pelo seguro para RP-CME, sendo usados fora das indicações aprovadas.
Cirurgia de catarata: Realizada em pacientes com catarata subcapsular posterior. Largura da EZ ≥600 μm na OCT pré-operatória é um fator preditivo de boa acuidade visual pós-operatória 5). Em casos de fragilidade das zônulas de Zinn, considere o uso de anel de expansão capsular. Para prevenção de CME pós-operatória (10-14%), use colírios de esteroides e AINEs por período mais longo que o habitual 9).
Membrana epirretiniana (GL2026 CQ4): Vitrectomia. Melhora visual pode ser esperada em casos com linha EZ contínua. Em casos com linha EZ descontínua, a recuperação é limitada. Atrofia macular grave de longo prazo foi relatada, sendo recomendável avaliação em centro especializado 9).
Buraco macular: A vitrectomia é o único tratamento curativo. Estudos sobre resultados pós-operatórios são limitados 9).
Apoio e Reabilitação
Cuidados de baixa visão: Baixa visão → lupas, ampliadores de leitura, tablets; fotofobia → óculos de proteção; estreitamento de campo visual → bengala branca; visão de longe → monóculo; óculos auxiliares para visão noturna. Suporte individualizado conforme campo visual e acuidade é importante. Recomenda-se o uso do Smart Site (apresentação de centros de consulta de baixa visão em cada região).
Aconselhamento genético: Realizado por especialista em genética clínica e conselheiro genético certificado. Inclui estimativa de risco de recorrência, discussões sobre educação, emprego, casamento e gravidez. O aconselhamento genético pode ser recebido mesmo sem diagnóstico genético.
Sistema de doenças raras: O sistema de auxílio financeiro para doenças raras designadas está disponível 9). Considere também obter o cartão de deficiência física e apoio para tratamento autônomo.
Voretigene neparvovec (Luxturna): Medicamento de terapia gênica administrável a pacientes com variantes patogênicas bialélicas no gene RPE65 e células retinianas viáveis suficientes. Aprovado no Japão em 2023 9). No estudo de Fase III dos EUA (estudo 301), 31 pacientes foram inscritos, e a análise mITT (20 intervenção, 9 controle) mostrou melhora significativa no MLMT e FST de luz branca em relação ao grupo controle 18). No estudo de Fase III doméstico (estudo A11301), aumento da sensibilidade FST e expansão do campo visual foram confirmados em 4 pacientes japoneses 19). O efeito na melhora da acuidade visual é limitado, e atrofia coriorretiniana como complicação de longo prazo foi relatada em mais de 20% 9).
Realizar a cada 6 meses a 1 ano: acuidade visual, lâmpada de fenda, fundo de olho, perimetria de Humphrey (HFA 10-2), OCT9).
QQuais medicamentos são usados para tratar o edema macular?
A
A primeira linha de tratamento para RP-CME são os inibidores da anidrase carbônica, como colírio de dorzolamida ou comprimidos orais de acetazolamida9). A melhora da CMT é alcançada em cerca de 40% dos casos, mas cerca de 30% apresentam recorrência. Se não houver melhora com CAI, a injeção intravítrea de triancinolona acetonida ou o implante intravítreo de dexametasona (Ozurdex) são opções. Os anti-VEGF não são recomendados para RP-CME. Todos esses são usos off-label.
6. Fisiopatologia e mecanismos detalhados de ocorrência
A via final comum da morte dos fotorreceptores na RP é a apoptose. Embora os tipos de mutações genéticas sejam diversos, eles convergem para uma via comum de morte celular.
Na RP, primeiro os bastonetes degeneram e desaparecem, seguidos pela degeneração secundária dos cones 7). Os cones dependem de um fator trófico produzido pelos bastonetes (RdCVF) para sobreviver, portanto, após a perda dos bastonetes, os cones também perdem sua função 7)11).
A retina é um dos tecidos com maior atividade metabólica, convertendo 80-90% da glicose em lactato por glicólise aeróbica (efeito Warburg). Os cones são mais vulneráveis ao estresse metabólico do que os bastonetes, e essa vulnerabilidade metabólica também contribui para a degeneração secundária dos cones 11).
A inflamação também é reconhecida como um fator importante na progressão da RP, com ativação da microglia e infiltração de macrófagos agravando o dano retiniano 11). O estresse oxidativo também atua como um impulsionador biológico da degeneração secundária dos cones.
O mecanismo de degeneração difere conforme o gene.
Mutação RHO: A rodopsina mal dobrada induz estresse do retículo endoplasmático → resposta a proteínas mal dobradas → apoptose11)
Mutações REEP6: REEP6 codifica uma proteína envolvida na manutenção da morfologia do RE. Mutações patogênicas levam à formação de inclusões no RE no segmento externo dos bastonetes, resultando em degeneração fotorreceptora4)
Mutações RPGR: RPGR está envolvido na estrutura do axonema dos cílios primários, e mutações prejudicam o transporte de material para o segmento externo dos fotorreceptores1)
7. Pesquisas Recentes e Perspectivas Futuras (Relatos em Fase de Pesquisa)
A terapia gênica é a abordagem mais promissora para tratar doenças hereditárias da retina8).
Luxturna (voretigene neparvovec): Medicamento de terapia gênica administrável a pacientes com variantes patogênicas bialélicas no gene RPE65. No Fase III dos EUA (estudo 301), 31 pacientes foram inscritos, e a análise mITT (20 intervenção, 9 controle) mostrou melhora significativa no MLMT e FST de luz branca em comparação ao grupo controle18). No Fase III doméstico (estudo A11301), aumento da sensibilidade FST e expansão do campo visual foram confirmados em 4 japoneses19). Foi aprovado no Japão em 2023, servindo como ponte entre tratamento padrão e tratamento experimental.
Terapia gênica RPGR: A terapia gênica mediada por AAV para XL-RP devido a mutações RPGR avançou para ensaios clínicos de Fase I/II/III8).
CRISPR/Cas9: Pesquisas estão em andamento para corrigir diretamente mutações patogênicas ou inativar mutações dominantes negativas8).
RdCVF (Fator de Sobrevivência de Cone Derivado de Bastonete) e Terapia de Proteção de Cone
RdCVF é uma proteína secretada pelos bastonetes que mantém a sobrevivência dos cones7)11). Ensaios clínicos de terapia de proteção de cone usando RdCVF estão em andamento, sendo uma estratégia terapêutica independente para manter a função dos cones após degeneração dos bastonetes.
NAC é um medicamento que suprime o estresse oxidativo, e um ensaio de Fase I relatou melhora na acuidade visual17). Em 2025, um ensaio de Fase III está em andamento.
Potencial de Reposicionamento de Glicocorticoide (Dexametasona)
Um estudo in vivo recente (modelo de camundongo rd10) demonstrou que a dexametasona intravítrea protege os fotorreceptores cones e o epitélio pigmentar da retina11). Os glicocorticoides têm forte potencial de reposicionamento como terapia independente de mutação. No entanto, as evidências atuais são limitadas a modelos animais, sendo necessária maior verificação para aplicação clínica em humanos.
Transplante de retina derivada de células iPS e retina artificial
Transplante de retina derivada de células iPS: Pesquisas sobre o transplante de folhas de fotorreceptores feitas a partir das próprias células iPS do paciente estão em andamento.
Retina artificial (prótese de retina): Dispositivos de estimulação elétrica para RP em estágio avançado. O Argus II e outros foram colocados em prática no exterior, e ensaios clínicos do método de estimulação transcoroidal-retiniana estão em andamento no Japão.
QA terapia genética também está disponível no Japão?
A
O voretigene neparvovec (Luxturna) foi aprovado no Japão em 2023, mas é limitado a distrofias retinianas com variantes patogênicas bialélicas no gene RPE65 9)18)19). A terapia genética para RP com outras mutações genéticas, incluindo a mutação RPGR, ainda está em fase de ensaios clínicos 7) e não é aprovada como terapia geral no Japão.
QComo obter terapia em fase de pesquisa?
A
A participação em ensaios clínicos é limitada a ensaios oficiais aprovados pelo comitê de ética da instituição médica. Além de consultar o médico responsável, as informações sobre ensaios podem ser pesquisadas no site de Informações de Ensaios Clínicos (jRCT) operado pelo Centro Nacional do Câncer, ou no clinicaltrials.gov dos EUA.
Baz-Redón N, Sánchez-Bellver L, Fernández-Cancio M, Rovira-Amigo S, Burgoyne T, Ranjit R, Aquino V, Toro-Barrios N, et al. Primary Ciliary Dyskinesia and Retinitis Pigmentosa: Novel RPGR Variant and Possible Modifier Gene. Cells. 2024;13(6). doi:10.3390/cells13060524. PMID:38534367; PMCID:PMC10968961.
Holanda IP, et al. Syndromic Retinitis Pigmentosa: A 15-Patient Study Exploring Clinical and Genetic Features. Genes. 2024;15(4):516.
Wawrocka A, Walczak-Sztulpa J, Kuszel L, Niedziela-Schwartz Z, Skorczyk-Werner A, Bernardczyk-Meller J, Krawczynski MR. Coexistence of Retinitis Pigmentosa and Ataxia in Patients with PHARC, PCARP, and Oliver-McFarlane Syndromes. International journal of molecular sciences. 2024;25(11). doi:10.3390/ijms25115759. PMID:38891946; PMCID:PMC11172263.
Lujia Zhang, Ya Li, Litao Qin, Yu Wu, Bo Lei. Autosomal Recessive Retinitis Pigmentosa Associated with Three Novel REEP6 Variants in Chinese Population. Genes. 2021;12(4):537. doi:10.3390/genes12040537.
Daiki Sakai, Seiji Takagi, Yasuhiko Hirami, Makoto Nakamura, Yasuo Kurimoto. Use of ellipsoid zone width for predicting visual prognosis after cataract surgery in patients with retinitis pigmentosa. Eye. 2022;37(1):42-47. doi:10.1038/s41433-021-01878-3.
日本眼科学会. 網膜色素変性診療ガイドライン. 日眼会誌. 2016;120:846-861.
Campochiaro PA, Mir TA. The mechanism of cone cell death in Retinitis Pigmentosa. Prog Retin Eye Res. 2018;62:24-37. doi:10.1016/j.preteyeres.2017.08.004.
Liu Y, Zong X, Cao W, Zhang W, Zhang N, Yang N. Gene therapy for retinitis pigmentosa: current challenges and new progress. Biomolecules. 2024;14(8):903. PMID: 39199291. PMCID: PMC11352491. doi:10.3390/biom14080903.
Napoli D, Di Marco B, Salamone G, Orsini N, Mazziotti R, Strettoi E. Keeping the lights on: a new role for an old drug to support cone survival in Retinitis Pigmentosa. Progress in retinal and eye research. 2025;109:101403. doi:10.1016/j.preteyeres.2025.101403. PMID:40998215.
Koyanagi Y, Akiyama M, Nishiguchi KM, Momozawa Y, Kamatani Y, Takata S, et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. Journal of medical genetics. 2019;56(10):662-670. doi:10.1136/jmedgenet-2018-105691. PMID:31213501.
Numa S, Oishi A, Higasa K, Oishi M, Miyata M, Hasegawa T, Ikeda HO, Otsuka Y, Matsuda F, Tsujikawa A.. EYS is a major gene involved in retinitis pigmentosa in Japan: genetic landscapes revealed by stepwise genetic screening. Sci Rep. 2020;10(1):20770. doi:10.1038/s41598-020-77558-1. PMID:33247286; PMCID:PMC7695703.
Berson EL, Rosner B, Sandberg MA, Hayes KC, Nicholson BW, Weigel-DiFranco C, Willett W.. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761-772. doi:10.1001/archopht.1993.01090060049022. PMID:8512476.
Nakazawa M, Suzuki Y, Ito T, Metoki T, Kudo T, Ohguro H. Long-term effects of nilvadipine against progression of the central visual field defect in retinitis pigmentosa: an extended study. BioMed research international. 2013;2013:585729. doi:10.1155/2013/585729. PMID:24319686; PMCID:PMC3844269.
Yamamoto S, Sugawara T, Murakami A, Nakazawa M, Nao-I N, Machida S, et al. Topical isopropyl unoprostone for retinitis pigmentosa: microperimetric results of the phase 2 clinical study. Ophthalmology and therapy. 2012;1(1):5. doi:10.1007/s40123-012-0005-9. PMID:25135585; PMCID:PMC4108136.
Campochiaro PA, Iftikhar M, Hafiz G, Akhlaq A, Tsai G, Wehling D, et al. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. The Journal of clinical investigation. 2020;130(3):1527-1541. doi:10.1172/JCI132990. PMID:31805012; PMCID:PMC7269599.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Fujinami K, Akiyama K, Tsunoda K, et al. Efficacy and Safety of Voretigene Neparvovec in RPE65-Retinopathy: Results of a Phase III Trial in Japan. Ophthalmol Sci. 2025;5:100876. doi:10.1016/j.xops.2025.100876. PMID:40910102; PMCID:PMC12405627.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.