Épisclérite simple
Fréquence : plus courante
Début : brutal
Évolution : atteint un pic en environ 12 heures et disparaît en 2 à 3 jours
Signes : hyperhémie en éventail (environ 67 %) ou diffuse (environ 33 %)

L’épisclérite est une maladie congestive bénigne et auto-limitée du tissu épiscléral. Il s’agit d’une inflammation des plexus vasculaires superficiels, comme le plexus de la capsule de Tenon, avec une douleur moins intense et un impact visuel moindre que la sclérite qui affecte les vaisseaux plus profonds. La plupart des cas sont idiopathiques et récurrents, avec une tendance à l’atteinte bilatérale. L’incidence annuelle est de 41,0 pour 100 000 personnes et la prévalence de 52,6.
Bien qu’elle soit relativement fréquente parmi les causes de rougeur oculaire, elle peut être confondue avec une conjonctivite ou une sclérite, et est souvent mal diagnostiquée lors de la première consultation. Dans cette maladie, le parenchyme scléral lui-même n’est pas atteint, et l’évolution vers des complications structurelles graves comme la perforation oculaire est quasi inexistante. Cependant, dans les cas récurrents ou ceux associés à des maladies auto-immunes systémiques telles que la polyarthrite rhumatoïde ou la granulomatose avec polyangéite, un traitement de la maladie sous-jacente et un suivi à long terme sont nécessaires. Comprendre l’épisclérite non pas comme une maladie oculaire isolée mais comme une « manifestation oculaire » d’une maladie systémique est essentiel pour la gestion des récidives et l’amélioration du pronostic.
La classification de Watson est largement utilisée pour la classification clinique des maladies inflammatoires de la sclère et de l’épisclère. Selon la localisation, elles sont divisées en trois groupes : épisclérite, sclérite antérieure et sclérite postérieure. La sclérite antérieure est ensuite subdivisée en formes diffuse, nodulaire et nécrosante (inflammatoire/non inflammatoire) selon la morphologie. Une différence importante avec la sclérite antérieure est que l’épisclérite n’a pas de type nécrosant et est classée morphologiquement en deux types : simple (forme diffuse) et nodulaire. Cette classification reflète la profondeur de l’inflammation (superficielle ou profonde) et la gravité de l’évolution et du pronostic, de sorte que la détermination du type de maladie au moment du diagnostic constitue la base de la stratégie thérapeutique et de l’explication du pronostic. L’épisclérite est classée dans ce système comme le groupe le plus bénin et de meilleur pronostic.
Épisclérite simple
Fréquence : plus courante
Début : brutal
Évolution : atteint un pic en environ 12 heures et disparaît en 2 à 3 jours
Signes : hyperhémie en éventail (environ 67 %) ou diffuse (environ 33 %)
Épisclérite nodulaire
Fréquence : un peu moins fréquente
Début : progressif
Évolution : tendance à une durée plus longue des symptômes que l’épisclérite simple
Signes : nodule épiscléral localisé près du limbe cornéen (mobile)
La sclère est composée de trois couches : l’épisclère, le stroma scléral et la lamina fusca. L’épisclère est un tissu conjonctif contenant des vaisseaux situé au-dessus du stroma scléral, compris comme une structure fibro-élastique située entre le stroma scléral et la capsule de Tenon. Elle se compose de deux couches : la couche pariétale externe (réseau capillaire épiscléral superficiel) et la couche viscérale profonde (réseau vasculaire hautement anastomosé), toutes deux dérivées des artères ciliaires antérieures. La plupart des fibres nerveuses sont des branches du nerf trijumeau. L’épisclère forme un plexus vasculaire épiscléral entre les insertions des muscles droits et le limbe, normalement caché par la conjonctive, mais en cas d’inflammation, il se dilate et provoque une hyperhémie vive. L’épisclère s’amincit progressivement vers l’arrière du globe oculaire, où la capsule de Tenon devient prédominante.
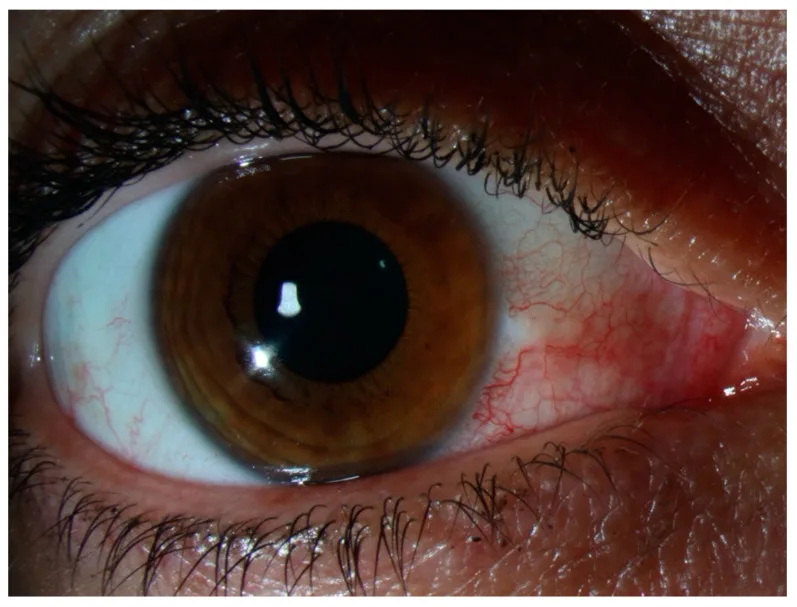
Il n’y a pas de douleur à la pression ni de sécrétion oculaire. En cas de douleur intense ou de sécrétion oculaire évidente, il faut reconsidérer une sclérite, une conjonctivite infectieuse ou une uvéite antérieure. Les symptômes disparaissent souvent en quelques jours ou complètement, sans affecter la fonction visuelle. En cas de récidive, elle survient souvent au même endroit ou à l’œil controlatéral, et le patient la remarque souvent comme un « œil rouge habituel ». Une douleur intense perturbant le sommeil nocturne comme dans la sclérite ou une forte douleur à la pression de la paupière supérieure ne sont généralement pas observées dans l’épisclérite.
L’observation de la localisation et de la couleur de la rougeur est essentielle pour le diagnostic différentiel. La rougeur de l’épisclérite est rouge vif à rose, contrastant avec la rougeur profonde rouge foncé (teinte violacée) observée dans la sclérite.
| Signe | Épisclérite | Sclérite |
|---|---|---|
| Couleur de la rougeur | Rouge vif à rose | Rouge foncé (violacé) |
| Douleur | Légère à absente | Forte et irradiante |
| Mobilité du nodule | Présente | Absente |
L’acuité visuelle est généralement normale. L’œdème conjonctival, l’hypertension oculaire, l’uvéite antérieure et la kératite sont rares ; leur présence doit faire évoquer une sclérite ou une autre pathologie. L’absence de signes inflammatoires de la conjonctive palpébrale est utile pour différencier de la conjonctivite. Dans la sclérite, l’inflammation peut s’étendre aux tissus adjacents, entraînant des infiltrations ou ulcérations cornéennes périphériques et une uvéite antérieure, alors que l’épisclérite est auto-limitée et n’implique presque jamais les tissus voisins. À la lampe à fente, on identifie le niveau du plexus vasculaire scléral ; si une lésion rouge surélevée ne permet pas de voir les vaisseaux scléraux, il faut envisager une lésion tumorale.
L’épisclérite se caractérise par l’absence de sécrétions oculaires et une hyperhémie localisée près du limbe cornéen. La conjonctivite est généralement indolore, avec des sécrétions, et l’hyperhémie est plus marquée au niveau du fornix, diminuant vers le limbe. À la lampe à fente, les vaisseaux épiscléraux ne sont pas mobiles, contrairement aux vaisseaux conjonctivaux. Voir la section « Diagnostic et méthodes d’examen » pour plus de détails.
La majorité des cas sont idiopathiques (cause inconnue), avec une association à une maladie systémique rapportée dans environ 26 à 36 % des cas. Même idiopathique, un mécanisme immunologique est suggéré, reposant sur une réaction inflammatoire non spécifique à prédominance lymphocytaire dans le plexus vasculaire épiscléral superficiel. L’évolution récurrente et la tendance à l’atteinte bilatérale suggèrent un trouble de la régulation immunitaire systémique sous-jacent.
Collagénoses et maladies auto-immunes (la plus fréquente étant la polyarthrite rhumatoïde)1) :
Vascularite :
Infections : les bactéries, mycobactéries, syphilis, maladie de Lyme, virus de l’herpès, zona, etc. peuvent en être la cause. L’épisclérite associée au zona ophtalmique est considérée comme une réponse immunitaire à l’agent pathogène plutôt qu’une infection directe. Un cas de parasitose sous-conjonctivale à Dirofilaria repens a été diagnostiqué à tort comme une épisclérite7).
Autres : goutte, atopie, corps étranger, traumatisme chimique, médicaments (topiramate, pamidronate), et signalée comme symptôme initial de la COVID-19.
Oui, environ 30 % des patients présentent une maladie systémique associée. La plus fréquente est la polyarthrite rhumatoïde, mais elle peut aussi être le premier symptôme de maladies comme la granulomatose avec polyangéite (GPA) ou la maladie de Behçet, dont le pronostic dépend d’un diagnostic et d’un traitement précoces. En cas de récidives fréquentes ou de symptômes systémiques, un bilan systémique incluant facteur rhumatoïde, anticorps antinucléaires, ANCA et analyse d’urine est recommandé.
L’épisclérite est principalement un diagnostic clinique basé sur l’anamnèse et l’examen à la lampe à fente. L’observation minutieuse à la lampe à fente du niveau des vaisseaux scléraux (superficiel ou profond), de la teinte de l’hyperhémie, de la présence de nodules, et de l’amincissement ou de la nécrose est fondamentale.
L’instillation de phényléphrine à 2,5 % contracte les vaisseaux conjonctivaux et est utile pour différencier la conjonctivite de l’épisclérite. La phényléphrine à 10 % contracte le réseau vasculaire épiscléral superficiel mais pas le réseau profond, permettant de distinguer l’épisclérite de la sclérite.
Le test de réponse à l’instillation d’épinéphrine diluée au 1/1000 est une méthode simple pour évaluer l’implication des vaisseaux profonds. Si l’hyperhémie disparaît après instillation, cela suggère une épisclérite ; si elle persiste, une sclérite. L’évaluation globale combine le nombre et la mobilité des nodules, la présence de douleur et de sensibilité, et la réponse à l’épinéphrine.
Les tests de réponse à l’épinéphrine et à la phényléphrine sont particulièrement utiles comme aide diagnostique lorsque la structure en couches de l’hyperhémie ne peut pas être directement confirmée à la lampe à fente ou dans les cas de petits nodules. L’observation 10 à 15 minutes après instillation permet de juger de la contraction des vaisseaux superficiels ; si l’hyperhémie des vaisseaux profonds persiste, la priorité est donnée à la prise en charge d’une sclérite.
La ténonites est également considérée comme un type d’épisclérite, et la distinction clinique entre les deux est difficile. La décision repose sur une combinaison de la mobilité des nodules, de la présence de douleur/sensibilité, de la réponse aux gouttes d’épinéphrine et des résultats de la coloration à la fluorescéine.
Pour une épisclérite unique et légère, un bilan systémique étendu n’est pas nécessaire. En cas de récidives fréquentes ou de symptômes systémiques associés, envisager les examens suivants.
Dans les cas où une épisclérite apparaît comme première manifestation de la granulomatose avec polyangéite, une insuffisance rénale peut être associée 3). Lorsqu’une inflammation oculaire et une anomalie de la fonction rénale sont toutes deux présentes, il convient de rechercher rapidement une vascularite systémique, y compris la granulomatose avec polyangéite. En cas d’épisclérite réfractaire ou récurrente, il est souhaitable d’évaluer l’activité de la maladie et d’initier un traitement de la maladie sous-jacente en collaboration avec un rhumatologue ou un interniste.
En plus de l’évaluation par lampe à fente, l’épaisseur de la couche épisclérale et le trajet vasculaire peuvent être évalués par tomographie par cohérence optique du segment antérieur (AS-OCT), et l’épaisseur sclérale par échographie (mode B) comme aide au diagnostic. Pour exclure une sclérite nécrosante ou évaluer la présence d’une sclérite postérieure, l’échographie vérifie la présence d’un signe T (épanchement périscléral). Dans l’épisclérite typique, ces examens d’imagerie manquent souvent de signes spécifiques, et le diagnostic repose sur une combinaison d’examen direct à la lampe à fente, d’anamnèse et de bilan systémique.
L’épisclérite guérit souvent spontanément en quelques jours à quelques semaines sans traitement. La première étape de la prise en charge consiste à expliquer au patient la nature bénigne et l’évolution naturelle de la maladie, ainsi que la nécessité de rechercher une maladie systémique, et à le rassurer. Les compresses froides et les larmes artificielles réfrigérées sont efficaces pour réduire les symptômes subjectifs tels que l’irritation et la sensation de chaleur. Dans les cas bénins, une intervention médicamenteuse agressive n’est pas nécessaire ; une courte surveillance de quelques jours pour confirmer la résolution spontanée permet d’éviter les rebonds et les effets secondaires liés au traitement.
Les collyres corticostéroïdes à faible concentration sont le traitement de première intention. Un collyre antibiotique est souvent associé pour aider à différencier une sclérite et prévenir une surinfection.
Si la réponse au traitement par collyre est insuffisante, il faut envisager de passer à l’examen et au traitement d’une sclérite. Bien que les collyres corticostéroïdes soulagent rapidement les symptômes, une utilisation prolongée ou répétée peut augmenter le risque de récidive et provoquer une hyperhémie de rebond.
Le traitement repose sur une réduction progressive et un arrêt après la disparition des symptômes, en évitant une administration continue prolongée. L’utilisation à long terme de collyres stéroïdiens comporte un risque d’augmentation de la pression intraoculaire réactive aux stéroïdes et de cataracte sous-capsulaire postérieure ; il convient donc de réduire progressivement après confirmation de l’amélioration dans un délai de 1 à 2 semaines. En cas de récidive, évaluer individuellement l’activité de la maladie lors de chaque épisode et privilégier l’optimisation du traitement de la maladie systémique sous-jacente.
Dans l’épisclérite associée à des maladies du collagène telles que la polyarthrite rhumatoïde, le traitement de la maladie sous-jacente a un impact direct sur le pronostic1). En cas de résistance au traitement local, associer une corticothérapie orale par prednisolone (20 à 30 mg/jour avec réduction progressive). Sauf en cas d’évidence d’une maladie inflammatoire systémique associée, les cas nécessitant une corticothérapie systémique sont très rares.
Dans l’épisclérite associée à la granulomatose avec polyangéite, un traitement d’induction de rémission par cyclophosphamide ou rituximab est efficace3)4). Le rituximab a montré un taux de rémission à 6 mois plus élevé que le cyclophosphamide (64 % contre 53 %) selon certaines études3).
Les collyres stéroïdiens suppriment rapidement les symptômes de l’épisclérite, mais il a été signalé qu’ils peuvent provoquer une rougeur par « rebond » après l’arrêt, entraînant une rechute plus sévère. Par conséquent, l’utilisation de stéroïdes est controversée ; dans les cas bénins, certains préfèrent une observation sans traitement ou privilégient les AINS. En cas de récidives fréquentes, il est recommandé d’envisager un inhibiteur de la COX-2 par voie orale ou un bilan de la maladie systémique.
Le mécanisme de l’épisclérite n’est pas encore complètement élucidé. Dans la zone lésée, on observe une dilatation et une congestion du réseau vasculaire épiscléral superficiel, avec une infiltration de cellules inflammatoires, principalement des lymphocytes, dans l’épisclère et la capsule de Tenon. La différence essentielle avec la sclérite est que le parenchyme scléral lui-même n’est pas atteint. L’infiltrat inflammatoire est principalement composé de cellules T et de quelques plasmocytes ; on n’observe généralement pas d’image de suppuration à prédominance neutrophile ni de formation de granulomes.
Histologiquement, il s’agit d’une inflammation non granulomateuse, caractérisée principalement par une dilatation vasculaire et une infiltration lymphocytaire. Dans l’épisclérite nodulaire, on observe au centre de la lésion une nécrose fibrinoïde entourée d’un arrangement de cellules épithélioïdes. Ces résultats sont similaires à ceux de l’inflammation granulomateuse observée dans la sclérite, et certains considèrent l’épisclérite et la sclérite comme un spectre basé sur la profondeur de l’inflammation. La petite nécrose fibrinoïde observée dans l’épisclérite peut être comprise comme une forme bénigne des changements nécrotiques plus étendus de la sclérite.
La progression de l’inflammation augmente la production d’espèces réactives de l’oxygène (ROS) et exacerbe le stress oxydatif2). La quantité totale de vitamine C dans la rétine humaine est environ 20 fois plus élevée que dans le plasma, et les tissus oculaires dépendent fortement du système antioxydant. Dans l’épisclérite auto-immune, il a été suggéré qu’une diminution de la fonction de ce système antioxydant pourrait provoquer une inflammation chronique et des lésions tissulaires de l’épisclère2). Les ROS endommagent l’endothélium vasculaire et induisent la libération de cytokines inflammatoires, provoquant une vasodilatation persistante et une augmentation de la perméabilité. L’exposition chronique au stress oxydatif de la surface oculaire et de l’épisclère est considérée comme un facteur contributif à l’épisclérite récurrente, et l’intérêt thérapeutique d’une intervention antioxydante est à l’étude.
Cliniquement, l’épisclérite se transforme rarement directement en sclérite. En revanche, la plupart des sclérites présentent également une inflammation de l’épisclère (changements épiscléritiques), de sorte que les deux sont comprises non pas comme des maladies complètement indépendantes, mais comme un continuum basé sur la profondeur de l’inflammation des couches vasculaires. L’épisclérite affecte principalement le réseau vasculaire épiscléral superficiel (couche pariétale), tandis que la sclérite affecte le réseau vasculaire profond et le parenchyme scléral.
Au niveau de l’insertion du muscle droit, l’épaisseur de la sclère est d’environ 0,3 mm, ce qui est le plus fin, et on sait qu’elle est plus vulnérable à l’inflammation et aux traumatismes. Le plexus vasculaire épiscléral reçoit un apport sanguin abondant via les artères ciliaires antérieures, de sorte que l’hyperémie se manifeste rapidement lors de l’inflammation. En revanche, la sclère elle-même est un tissu pauvre en vaisseaux, et une inflammation profonde comme la sclérite est rare. La caractéristique anatomique selon laquelle les vaisseaux provenant des artères ciliaires antérieures sont réversiblement congestionnés dans l’épisclérite est la base mécanistique de la disparition rapide de l’hyperémie lors du test à l’épinéphrine ; l’absence de cette réaction dans la vascularite sclérale profonde constitue une base physiopathologique pour le diagnostic différentiel.
Un rapport de cas décrit un homme de 60 ans atteint d’épisclérite idiopathique récurrente qui n’a présenté aucune récidive pendant 7 mois après avoir commencé à prendre 500 mg/jour de vitamine C par voie orale2). La vitamine C est un puissant antioxydant et il a été suggéré qu’elle pourrait supprimer l’inflammation des tissus oculaires en réduisant le stress oxydatif. On sait que les tissus oculaires dépendent fortement du système antioxydant, la concentration de vitamine C dans la rétine atteignant environ 20 fois celle du plasma, et la supplémentation en vitamine C et autres nutriments antioxydants pourrait être une stratégie candidate pour prévenir les récidives2). Cependant, des études cas-témoins contrôlées et des essais cliniques sont nécessaires pour établir son efficacité2). À l’heure actuelle, elle n’est envisagée qu’à titre adjuvant dans les cas de récidive sévère ou ceux associés à une sécheresse oculaire ou une inflammation chronique de la surface oculaire.
La granulomatose avec polyangéite (GPA) est une maladie mortelle avec un taux de mortalité à un an de 80 % si elle n’est pas traitée, mais l’introduction d’un traitement immunosuppresseur peut réduire la mortalité à 10 %3). Comme l’épisclérite peut être le premier symptôme de la GPA, les ophtalmologistes doivent reconnaître cette association et pratiquer activement des examens systémiques en cas d’épisclérite récurrente3)4). En particulier, la coexistence d’une inflammation oculaire et d’une insuffisance rénale est un signe fortement évocateur de granulomatose avec polyangéite3).
L’efficacité des agents biologiques tels que les inhibiteurs du TNFα et le rituximab a été rapportée dans l’épisclérite et la sclérite associées à la polyarthrite rhumatoïde1). L’infliximab et l’adalimumab ont fait leurs preuves dans la polyarthrite rhumatoïde et l’uvéite, et leur application est envisagée dans la sclérite et l’épisclérite réfractaires. En revanche, l’étanercept est connu pour provoquer ou aggraver une inflammation oculaire par une réaction paradoxale, ce qui nécessite une prudence dans le choix du médicament1). Le rituximab est un anticorps monoclonal ciblant les lymphocytes B, et son efficacité a été suggérée dans l’inflammation oculaire liée aux vascularites. L’utilisation de ces agents biologiques est décidée en étroite collaboration avec les services de rhumatologie et de médecine interne des maladies auto-immunes.
Des cas ont été rapportés où des patients diagnostiqués avec une épisclérite présentaient en réalité une tumeur métastatique intraoculaire 6) ou une parasitose sous-conjonctivale 7), soulignant l’importance d’exclure une maladie maligne ou infectieuse dans les épisclérites réfractaires ou récurrentes. L’imagerie et l’évaluation détaillée à la lampe à fente des masses incluant les vaisseaux sont des indices diagnostiques. Il est nécessaire d’évaluer la mobilité de la masse, la transparence des vaisseaux scléraux, la présence d’adhérences aux tissus environnants et la réponse au traitement. Une lésion surélevée persistante ne répondant pas aux corticostéroïdes topiques habituels justifie une biopsie et un examen d’imagerie approfondi.
Les études observationnelles à long terme sur l’évolution naturelle de l’épisclérite et le délai d’apparition des maladies systémiques sont limitées, et les données sur l’incidence et le profil des maladies associées dans la population japonaise sont insuffisantes. Les rapports antérieurs provenant d’Europe et des États-Unis indiquent une incidence annuelle d’environ 40 à 60 cas pour 100 000 personnes, mais les chiffres varient en fonction de l’ethnie, de l’environnement de vie et des différences dans les registres d’uvéite. La mise en place future de registres cliniques et d’études multicentriques devrait permettre d’identifier les facteurs de risque de récidive et le calendrier d’apparition des maladies systémiques.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.