Epiescleritis simple
Frecuencia: más común
Inicio: repentino
Curso: alcanza su punto máximo en aproximadamente 12 horas y desaparece en 2-3 días
Hallazgos: enrojecimiento en forma de abanico (aproximadamente 67%) o difuso (aproximadamente 33%)

La episcleritis es una enfermedad congestiva benigna y autolimitada del tejido epiescleral. Es una inflamación de los plexos vasculares superficiales, como el plexo de la cápsula de Tenon, y en comparación con la escleritis, que afecta vasos más profundos, el dolor es leve y el impacto en la visión es mínimo. En su mayoría es idiopática y recurrente, con tendencia a presentarse en ambos ojos. La incidencia reportada es de 41.0 casos por 100,000 personas al año, con una prevalencia de 52.6.
Aunque es una causa relativamente frecuente de enrojecimiento ocular, a menudo se confunde con conjuntivitis o escleritis, y no es raro que se diagnostique erróneamente en la primera consulta. En esta enfermedad, el parénquima escleral no se ve afectado y casi nunca progresa a complicaciones estructurales graves como la perforación ocular. Sin embargo, en casos con curso recurrente o en aquellos con enfermedades autoinmunes sistémicas subyacentes como artritis reumatoide o granulomatosis con poliangeítis, se requiere tratamiento de la enfermedad de base y seguimiento a largo plazo. Entenderla no solo como una enfermedad ocular aislada, sino como una “manifestación ocular” de una enfermedad sistémica, es clave para el manejo de las recurrencias y la mejora del pronóstico.
La clasificación de Watson se utiliza ampliamente para la clasificación clínica de las enfermedades inflamatorias de la esclera y la epiesclera. Se divide en tres grupos según la ubicación: epiescleritis, escleritis anterior y escleritis posterior. La escleritis anterior se subdivide además según la morfología en difusa, nodular y necrotizante (inflamatoria/no inflamatoria). Una diferencia importante con la escleritis anterior es que la epiescleritis no tiene un tipo necrotizante y se clasifica morfológicamente en dos tipos: simple (difusa) y nodular. Esta clasificación refleja la profundidad de la inflamación (superficial o profunda) y la gravedad del curso y pronóstico, por lo que la determinación del tipo de enfermedad en el momento del diagnóstico es la base para el plan de tratamiento y la explicación del pronóstico. La epiescleritis se clasifica como el grupo más leve y de mejor pronóstico dentro de esta clasificación.
Epiescleritis simple
Frecuencia: más común
Inicio: repentino
Curso: alcanza su punto máximo en aproximadamente 12 horas y desaparece en 2-3 días
Hallazgos: enrojecimiento en forma de abanico (aproximadamente 67%) o difuso (aproximadamente 33%)
Epiescleritis nodular
Frecuencia: algo menos común
Inicio: gradual
Curso: tiende a prolongarse más que la simple
Hallazgos: nódulo epiescleral localizado cerca del limbo corneal (móvil)
La esclera se compone de tres capas: epiesclera, estroma escleral y lámina fusca. La epiesclera es un tejido conectivo que contiene vasos sanguíneos sobre el estroma escleral y se entiende como una estructura fibroelástica ubicada entre el estroma escleral y la cápsula de Tenon. Consta de dos capas: la capa parietal externa (red capilar epiescleral superficial) y la capa visceral profunda (red vascular altamente anastomosada), ambas derivadas de las arterias ciliares anteriores. La mayoría de las fibras nerviosas son ramas del nervio trigémino. La epiesclera forma un plexo vascular epiescleral entre la inserción del músculo recto y el limbo, normalmente oculto por la conjuntiva, pero cuando se inflama, se dilata y produce un enrojecimiento brillante. La epiesclera se vuelve gradualmente más delgada hacia la parte posterior del ojo, donde la cápsula de Tenon se vuelve predominante.
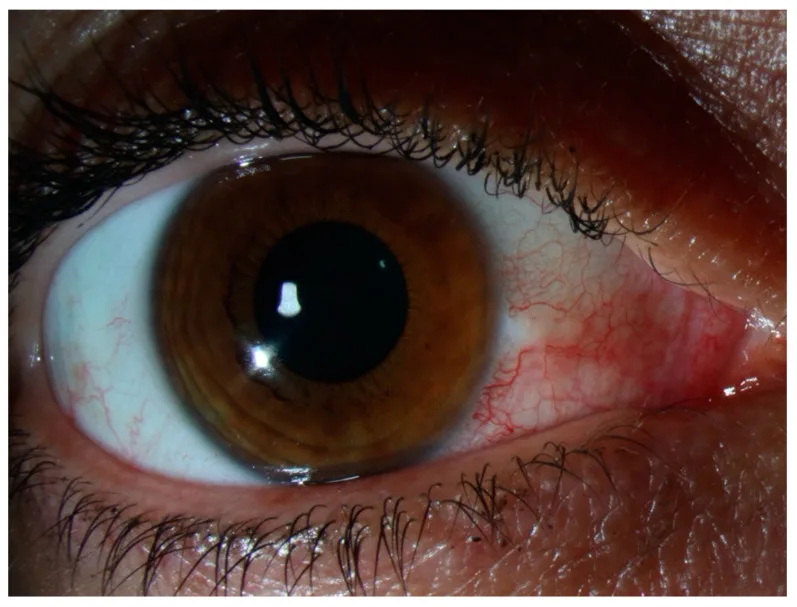
No hay dolor a la palpación ni secreción ocular. Si hay dolor intenso o secreción evidente, se debe reconsiderar escleritis, conjuntivitis infecciosa o uveítis anterior. Los síntomas suelen mejorar o desaparecer por completo en pocos días, sin afectar la función visual. En las recurrencias, suele aparecer en la misma zona o en el ojo contralateral, y los pacientes a menudo lo reconocen como «el ojo rojo de siempre». El dolor intenso que interrumpe el sueño nocturno, como en la escleritis, o el dolor fuerte a la palpación del párpado superior, no suele estar presente en la epiescleritis.
La observación de la localización y el color de la hiperemia es clave para el diagnóstico diferencial. La hiperemia en la epiescleritis es de color rojo brillante a rosado, en contraste con la hiperemia profunda de color rojo oscuro (tono violáceo) que se observa en la escleritis.
| Hallazgo | Epiescleritis | Escleritis |
|---|---|---|
| Color de la hiperemia | Rojo brillante a rosado | Rojo oscuro (violáceo) |
| Dolor | Leve a ninguno | Intenso y radiante |
| Movilidad del nódulo | Presente | Ausente |
La agudeza visual generalmente es normal. El edema conjuntival, la hipertensión ocular, la uveítis anterior y la queratitis son complicaciones raras; si están presentes, se debe considerar escleritis u otras enfermedades. La ausencia de signos inflamatorios en la conjuntiva tarsal es útil para diferenciarla de la conjuntivitis. Mientras que la escleritis puede causar infiltrados corneales periféricos, úlceras y uveítis anterior debido a la propagación de la inflamación a los tejidos circundantes, la epiescleritis es autolimitada y casi nunca afecta tejidos adyacentes. Con la lámpara de hendidura, se identifica el nivel del plexo vascular escleral; si se observa una lesión elevada rojiza pero no se pueden visualizar los vasos esclerales, también se debe considerar la posibilidad de una lesión neoplásica.
La epiescleritis se caracteriza por la ausencia de secreción ocular y la localización del enrojecimiento cerca del limbo corneal. La conjuntivitis generalmente no duele y presenta secreción ocular, con enrojecimiento más prominente en el fondo de saco y que disminuye hacia el limbo. Con la lámpara de hendidura, los vasos epiesclerales no son móviles, mientras que los vasos conjuntivales sí lo son, otro punto de diferenciación. Consulte la sección «Diagnóstico y métodos de examen» para más detalles.
La mayoría son idiopáticas (causa desconocida), y se reporta que aproximadamente el 26-36% de todos los casos se asocian con enfermedades sistémicas. Incluso en los casos idiopáticos, se sugiere la participación de mecanismos inmunológicos, basados en una reacción inflamatoria inespecífica centrada en linfocitos en el plexo vascular epiescleral superficial. El curso recurrente y la tendencia a la afectación bilateral sugieren una desregulación inmunológica sistémica subyacente.
Enfermedades del colágeno y autoinmunes (la más frecuente es la artritis reumatoide)1):
Vasculitis:
Infecciones: bacterias, micobacterias, sífilis, enfermedad de Lyme, virus del herpes, herpes zóster, entre otros, pueden ser causas. La epiescleritis asociada al herpes zóster oftálmico se considera una respuesta inmune al patógeno más que una infección directa. También se ha reportado un caso de parasitosis subconjuntival por Dirofilaria repens que fue diagnosticado erróneamente como epiescleritis7).
Otros: gota, atopia, cuerpos extraños, traumatismo químico, fármacos (topiramato, pamidronato), y se ha reportado como síntoma inicial de COVID-19.
Sí, aproximadamente el 30% de los pacientes presentan una enfermedad sistémica asociada. La más frecuente es la artritis reumatoide, pero también puede ser el síntoma inicial de enfermedades como la granulomatosis con poliangeítis (GPA) o la enfermedad de Behçet, cuyo diagnóstico y tratamiento tempranos son cruciales para el pronóstico. En casos de recurrencia o síntomas sistémicos, se recomienda una evaluación sistémica que incluya factor reumatoide, anticuerpos antinucleares, ANCA y análisis de orina.
La epiescleritis se diagnostica principalmente mediante la historia clínica y el examen con lámpara de hendidura. Es fundamental observar cuidadosamente con la lámpara de hendidura el nivel de los vasos esclerales (superficial o profundo), el tono de la hiperemia, la presencia de nódulos y si hay adelgazamiento o necrosis.
La fenilefrina al 2.5% contrae los vasos conjuntivales y es útil para diferenciar la conjuntivitis de la epiescleritis. La fenilefrina al 10% contrae la red vascular epiescleral superficial pero no la profunda, lo que permite diferenciar la epiescleritis de la escleritis.
La prueba de respuesta con epinefrina diluida 1:1000 es un método sencillo para determinar la afectación de los vasos profundos. Si la hiperemia desaparece tras la instilación, sugiere epiescleritis; si no desaparece, sugiere escleritis. Se evalúa de forma integral combinando el número y la movilidad de los nódulos, la presencia de dolor y sensibilidad, y la respuesta a la epinefrina.
Las pruebas de respuesta con epinefrina y fenilefrina son especialmente útiles como ayuda diagnóstica cuando no se puede confirmar directamente la estructura de capas de la hiperemia con la lámpara de hendidura o en casos con nódulos pequeños. Se observa la contracción de los vasos superficiales a los 10-15 minutos de la instilación; si persiste la hiperemia de los vasos profundos, se prioriza el manejo de la escleritis.
La tenonitis también se considera un tipo de epiescleritis, y la diferenciación clínica entre ambas es difícil. Se evalúa combinando la movilidad del nódulo, la presencia de dolor y sensibilidad, la respuesta a la instilación de epinefrina y los hallazgos de la tinción con fluoresceína.
En la epiescleritis única y leve no es necesaria una búsqueda sistémica extensa. En casos de recurrencia o con síntomas sistémicos, considerar las siguientes pruebas.
En casos donde la epiescleritis aparece como manifestación inicial de la granulomatosis con poliangeítis, puede coexistir disfunción renal3). Si se observan tanto inflamación ocular como anomalías en la función renal, se debe realizar una búsqueda rápida de vasculitis sistémica, incluida la granulomatosis con poliangeítis. En la epiescleritis refractaria o recurrente, es recomendable evaluar la actividad de la enfermedad e iniciar el tratamiento de la enfermedad subyacente en colaboración con reumatología y medicina interna.
Además de la evaluación con lámpara de hendidura, la tomografía de coherencia óptica del segmento anterior (AS-OCT) para evaluar el grosor de la capa epiescleral y el trayecto vascular, y la ecografía (modo B) para evaluar el grosor escleral, pueden utilizarse como ayudas diagnósticas. Para descartar escleritis necrotizante o evaluar la presencia de escleritis posterior, se verifica la presencia del signo T (acumulación de líquido perineural del nervio óptico) en la ecografía. En la epiescleritis típica, estas pruebas de imagen a menudo carecen de hallazgos específicos, por lo que el diagnóstico se realiza mediante la combinación de la exploración directa con lámpara de hendidura, la anamnesis y la evaluación sistémica.
La epiescleritis suele curar espontáneamente en días o semanas sin tratamiento. Explicar al paciente la naturaleza benigna de la enfermedad, su curso natural y la necesidad de buscar enfermedades sistémicas, así como brindar tranquilidad, es el primer paso del manejo. Las compresas frías y las lágrimas artificiales refrigeradas son efectivas para reducir síntomas subjetivos como irritación y sensación de calor. En casos leves, se evita la intervención farmacológica activa y se confirma la mejoría espontánea con seguimiento a corto plazo de varios días, evitando así rebotes o efectos secundarios asociados al tratamiento.
El colirio de esteroides de baja concentración es la primera opción. A menudo se combina con colirio antibiótico para ayudar en el diagnóstico diferencial con la escleritis.
Si la respuesta al tratamiento con gotas es escasa, se debe considerar cambiar a la evaluación y tratamiento de la escleritis. Las gotas de esteroides suprimen rápidamente los síntomas, pero se ha señalado que su uso prolongado y repetido aumenta el riesgo de recurrencia y puede inducir enrojecimiento de “rebote”.
El tratamiento se basa en reducir gradualmente y suspender tras la remisión de los síntomas, evitando la administración continua sin criterio. El uso prolongado de colirios esteroideos conlleva riesgo de aumento de la presión ocular inducido por esteroides y cataratas subcapsulares posteriores, por lo que se debe reducir gradualmente tras confirmar mejoría en 1-2 semanas. En casos recurrentes, se evalúa individualmente la actividad de cada episodio y se prioriza optimizar el tratamiento de la enfermedad sistémica subyacente.
En la epiescleritis asociada a enfermedades del colágeno como la artritis reumatoide, el tratamiento de la enfermedad de base influye directamente en el pronóstico1). Si la terapia local es resistente, se añade prednisolona oral (régimen de reducción gradual desde 20-30 mg/día). Excepto cuando la inflamación sistémica es evidente, los casos que requieren esteroides sistémicos son muy raros.
En la epiescleritis asociada a granulomatosis con poliangitis, la terapia de inducción de remisión con ciclofosfamida o rituximab es eficaz3)4). Se ha reportado que rituximab tiene una tasa de remisión a los 6 meses mayor que ciclofosfamida (64% frente a 53%)3).
Los colirios esteroideos suprimen rápidamente los síntomas de la epiescleritis, pero se ha señalado que pueden causar enrojecimiento por “efecto rebote” al suspenderlos, provocando una recaída más intensa. Por ello, su uso es controvertido; en casos leves, algunos prefieren observación sin tratamiento o priorizar AINE. En recurrencias repetidas, se recomienda el uso de inhibidores de COX2 por vía oral o la evaluación de enfermedades sistémicas.
El mecanismo de la epiescleritis aún no se comprende completamente. En las áreas afectadas, se produce dilatación y congestión de los vasos epiesclerales superficiales, con infiltración de células inflamatorias, principalmente linfocitos, en la epiesclera y la cápsula de Tenon. La diferencia esencial con la escleritis es que el parénquima escleral en sí no se ve afectado. El infiltrado inflamatorio consiste principalmente en células T y algunos plasmocitos; no suele observarse inflamación purulenta con predominio de neutrófilos ni formación de granulomas.
Histopatológicamente, es una inflamación no granulomatosa, con predominio de dilatación vascular e infiltración linfocítica. En la epiescleritis nodular, se observa necrosis fibrinoide en el centro de la lesión, rodeada por una disposición de células epitelioides. Estos hallazgos son similares a los de la inflamación granulomatosa de la escleritis, y existe la opinión de que la epiescleritis y la escleritis deben considerarse como un espectro según la profundidad de la inflamación. La necrosis fibrinoide de pequeño tamaño observada en la epiescleritis puede entenderse como una forma leve de los cambios necróticos más extensos de la escleritis.
La progresión de la inflamación aumenta la producción de especies reactivas de oxígeno (ROS) y potencia el estrés oxidativo2). La cantidad total de vitamina C en la retina humana es aproximadamente 20 veces mayor que en el plasma, y el tejido ocular depende en gran medida del sistema antioxidante. En la epiescleritis autoinmune, se ha sugerido que la disfunción de este sistema antioxidante podría causar inflamación crónica y daño tisular en la epiesclera2). Las ROS dañan el endotelio vascular e inducen la liberación de citocinas inflamatorias, provocando vasodilatación persistente y aumento de la permeabilidad. La exposición crónica al estrés oxidativo en la superficie ocular y la epiesclera se considera un factor contribuyente a la epiescleritis recurrente, y se está investigando el valor terapéutico de la intervención antioxidante.
Clínicamente, la epiescleritis rara vez progresa directamente a escleritis. Sin embargo, dado que en la mayoría de los casos de escleritis también se observa inflamación epiescleral (cambios similares a la epiescleritis), ambas entidades no se consideran enfermedades completamente independientes, sino un continuo según la profundidad de la afectación vascular. La epiescleritis afecta principalmente la red vascular epiescleral superficial (capa parietal), mientras que la escleritis afecta desde la red vascular profunda hasta el parénquima escleral.
Se sabe que en la inserción de los músculos rectos, la esclerótica es más delgada (aproximadamente 0.3 mm) y más vulnerable a la inflamación y el traumatismo. El plexo vascular epiescleral recibe un abundante suministro sanguíneo a través de las arterias ciliares anteriores, por lo que la congestión se manifiesta rápidamente durante la inflamación. Por otro lado, la esclerótica en sí es un tejido pobre en vasos, y la inflamación profunda como la escleritis es rara. La característica anatómica de que los vasos derivados de las arterias ciliares anteriores se congestionan reversiblemente en la epiescleritis es la base mecanicista de la rápida desaparición de la congestión con la prueba de fenilefrina tópica; la ausencia de esta reacción en la vasculitis escleral profunda constituye un fundamento fisiopatológico para el diagnóstico diferencial.
Existe un informe de caso de un hombre de 60 años con epiescleritis idiopática recurrente que, tras iniciar la administración oral de 500 mg/día de vitamina C, no presentó recaídas durante 7 meses2). Se ha señalado que la vitamina C es un potente antioxidante que podría suprimir la inflamación del tejido ocular mediante la reducción del estrés oxidativo. Se sabe que el tejido ocular depende en gran medida del sistema antioxidante, con concentraciones de vitamina C en la retina que alcanzan aproximadamente 20 veces las del plasma, por lo que la suplementación con vitamina C y otros nutrientes antioxidantes podría ser una estrategia candidata para prevenir recaídas2). Sin embargo, para establecer su eficacia se necesitan estudios de casos y controles y ensayos clínicos con grupo de control2). En la etapa actual, solo se considera de forma complementaria en casos de recaídas con síntomas intensos o en aquellos con ojo seco subyacente o inflamación crónica de la superficie ocular.
La granulomatosis con poliangitis es una enfermedad mortal que, sin tratamiento, alcanza una tasa de mortalidad del 80% al año, pero con terapia inmunosupresora se puede reducir al 10%3). Dado que la epiescleritis puede ser el síntoma inicial de la GPA, los oftalmólogos deben reconocer esta asociación y realizar exámenes sistémicos de forma activa en casos de epiescleritis recurrente3)4). En particular, la coexistencia de inflamación ocular y disfunción renal es un hallazgo que sugiere fuertemente granulomatosis con poliangitis3).
Se ha informado de la eficacia de agentes biológicos como los inhibidores del TNFα y rituximab en la epiescleritis y escleritis asociadas a artritis reumatoide1). Infliximab y adalimumab tienen un historial comprobado en artritis reumatoide y uveítis, y se considera su aplicación en escleritis y epiescleritis refractarias. Por otro lado, se sabe que etanercept puede inducir o exacerbar la inflamación ocular como reacción paradójica, por lo que se requiere precaución en la selección del fármaco1). Rituximab es un anticuerpo monoclonal dirigido contra las células B, y se ha sugerido su eficacia en la inflamación ocular asociada a vasculitis. El uso de estos agentes biológicos se decide en estrecha colaboración con el servicio de reumatología o medicina de colágeno.
Se han reportado casos de pacientes diagnosticados con epiescleritis que en realidad presentaban un tumor metastásico intraocular 6) o una parasitosis subconjuntival 7), por lo que en la epiescleritis refractaria o recurrente es importante descartar enfermedades malignas o infecciosas. Las pruebas de imagen y la evaluación detallada de los hallazgos con lámpara de hendidura de masas que incluyen vasos proporcionan pistas diagnósticas. Se requiere una evaluación integral de la movilidad de la masa, la transparencia de los vasos esclerales, la presencia de adherencias a los tejidos circundantes y la respuesta al tratamiento; las lesiones elevadas persistentes que no responden al tratamiento habitual con gotas de esteroides justifican la consideración activa de biopsia y estudios de imagen.
Los estudios de observación a largo plazo sobre la evolución natural de la epiescleritis y el tiempo hasta la manifestación de enfermedades sistémicas son limitados, y los datos sobre la incidencia y el perfil de enfermedades concomitantes en la población japonesa son insuficientes. Los informes previos de Europa y Estados Unidos muestran una incidencia anual de aproximadamente 40 a 60 casos por cada 100,000 personas, pero las cifras varían según la etnia, el entorno de vida y las diferencias en el funcionamiento de los registros de uveítis. Se espera que la futura creación de registros clínicos y estudios multicéntricos permitan identificar factores de riesgo de recurrencia y el cronograma hasta la manifestación de enfermedades sistémicas.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.