La kératite ulcéreuse périphérique (peripheral ulcerative keratitis: PUK) est un groupe de maladies présentant une inflammation destructrice en forme de croissant du stroma cornéen près du limbe. Elle se caractérise par une dégénérescence du stroma cornéen, une infiltration de cellules inflammatoires et une perte épithéliale, et peut évoluer vers un amincissement cornéen, un descemétocèle et une perforation. Il s’agit d’un terme générique pour les maladies présentant un ulcère dans la région périphérique de la cornée, incluant l’ulcère de Mooren idiopathique (ulcère cornéen phagédénique), les ulcères associés aux collagénoses telles que la polyarthrite rhumatoïde, l’allergie staphylococcique (ulcère cornéen catarrhal), et la phlyctène cornéenne.
L’incidence est rare, de 0,2 à 3 cas par million de personnes par an, sans différence entre les sexes8). Environ 50 % des patients présentent une maladie systémique sous-jacente, dont environ 20 % sont infectieuses8). La polyarthrite rhumatoïde (PR) est la cause la plus fréquente, représentant 34 à 42 % des patients atteints de PUK2).
Contexte épidémiologique de la polyarthrite rhumatoïde
La polyarthrite rhumatoïde est la maladie la plus fréquente parmi les collagénoses. Au Japon, la prévalence est rapportée à 5,4 pour 1000 femmes et 1,1 pour 1000 hommes. Elle touche surtout les femmes de 30 à 60 ans, et la proportion d’hommes augmente avec l’âge. Environ 70 % des patients atteints de PR présentent une corrélation génétique avec HLA-DR4. On sait également que la PR peut se déclencher ou s’aggraver après un traumatisme ou un accouchement.
Parmi les formes de PR, celle compliquée de vascularite est appelée polyarthrite rhumatoïde maligne. Elle s’accompagne de sclérite, pleurésie, pneumonie interstitielle, péricardite, myocardite, mononévrite multiple, embolie de l’artère mésentérique et ulcères digitaux, avec un mauvais pronostic général. La plupart des PUK associées à la PR surviennent dans cette forme maligne, entraînant un amincissement cornéen rapide et une perforation.
La pathogenèse de la PUK impliquerait une réaction auto-immune contre les antigènes cornéens, le dépôt de complexes immuns circulants et une réaction d’hypersensibilité aux antigènes étrangers. La région limbique est particulière car elle est densément peuplée de vaisseaux sanguins, de systèmes immunitaire et nerveux, créant un environnement propice au dépôt de complexes immuns. Dans la PUK associée aux collagénoses, les auto-anticorps sériques se déposent dans le limbe et la périphérie cornéenne, déclenchant une réaction allergique de type III, et les enzymes de dégradation de la matrice extracellulaire libérées par les cellules immunitaires infiltrées contribuent à la formation de l’ulcère.
L’ulcère de Mooren, par définition, désigne un ulcère cornéen périphérique de cause inconnue et sans maladie auto-immune systémique associée. En revanche, la PUK survient dans le cadre d’une maladie systémique, et la distinction entre les deux est importante pour déterminer la stratégie thérapeutique. Dans l’ulcère de Mooren, la sclérite est légère, tandis que dans l’ulcère périphérique associé aux collagénoses, une lésion gris-blanc progresse vers le centre de la cornée, souvent accompagnée d’une invasion vasculaire, secondaire à une épisclérite ou une sclérite.
QQuelle est la différence entre la PUK et l'ulcère de Mooren ?
A
La PUK est un ulcère de la périphérie cornéenne survenant dans le cadre de maladies auto-immunes systémiques telles que la polyarthrite rhumatoïde ou les vascularites associées aux ANCA. En revanche, l’ulcère de Mooren est un ulcère marginal cornéen idiopathique sans maladie systémique, dont la pathogenèse impliquerait des auto-anticorps dirigés contre les cellules épithéliales cornéennes. Cliniquement, l’ulcère de Mooren s’accompagne d’une sclérite légère, tandis que la PUK est associée à une sclérite dans environ 36 % des cas. Le bilan systémique est la clé du diagnostic différentiel.
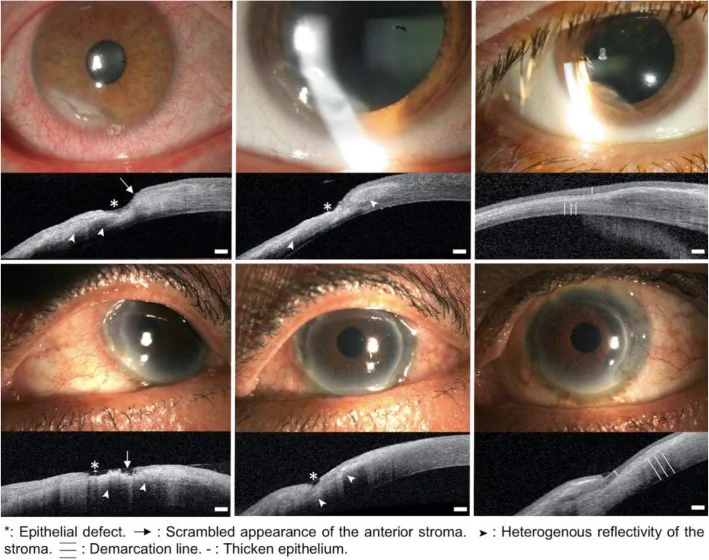
Bonnet C, Debillon L, Al-Hashimi S, et al. Anterior segment optical coherence tomography imaging in peripheral ulcerative keratitis, a corneal structural description. BMC Ophthalmol. 2020;20:205. Figure 1. PMCID: PMC7249626. License: CC BY 4.0.
Images à la lampe à fente et AS-OCT de deux cas ayant évolué favorablement sous traitement (en haut : premier cas, au milieu : deuxième cas), montrant de gauche à droite la phase active, le processus de guérison et la phase de guérison. Elles correspondent aux stades d’évolution des lésions (phase active, processus de guérison, phase de guérison) traités dans la section « 2. Principaux symptômes et signes cliniques ».
Douleur oculaire : Douleur intense associée à l’ulcère cornéen. En cas de sclérite associée, la douleur est profonde, irradiant vers le visage ou la tempe, et peut être suffisamment intense pour perturber le sommeil.
Hyperémie : accompagnée d’une injection ciliaire, souvent localisée. En cas de sclérite associée, on observe une hyperémie circonférentielle sévère due à la dilatation et à la tortuosité des vaisseaux scléraux.
Baisse de l’acuité visuelle : survient en fonction du degré d’opacité et d’amincissement cornéens.
Photophobie et larmoiement : apparaissent en raison d’une atteinte épithéliale cornéenne.
Sensation de sécheresse et de corps étranger : chez les patients atteints de polyarthrite rhumatoïde, un syndrome sec oculaire sévère (syndrome de Sjögren secondaire) est fréquemment associé, entraînant une sensation de corps étranger et de sécheresse à la surface oculaire.
Le signe cornéen de l’ulcère cornéen périphérique (PUK) est une destruction stromale en forme de croissant localisée près du limbe. Dans le PUK associé à une collagénose, la lésion progresse souvent vers le centre de la cornée après une sclérite ou une épisclérite.
Infiltration stromale et ulcération : on observe une infiltration et une nécrose stromale en forme de croissant parallèle au limbe. Le bord de l’ulcère présente un aspect de tunnel profond appelé undermining.
Perte épithéliale : l’épithélium de la zone ulcérée se détache et devient positif à la coloration à la fluorescéine.
Amincissement cornéen : en progressant, le stroma cornéen devient nettement aminci. Dans les cas sévères, on observe une cornée en sablier (hourglass cornea)4).
Perforation cornéenne paracentrale : dans les cas graves, une perforation se produit près du centre de la cornée. En cas de perforation, on observe une pupille en forme de poire et un prolapsus du tissu irien3).
Sclérite associée : environ 36 % des patients atteints de PUK développent une sclérite. La sclérite est une inflammation du plexus vasculaire scléral profond, provoquant un œdème et une infiltration cellulaire de la sclère.
Néovascularisation cornéenne : dans les cas chroniques, on observe une invasion vasculaire superficielle et profonde5).
Spectre des complications oculaires de la polyarthrite rhumatoïde
Les complications oculaires associées à la polyarthrite rhumatoïde présentent une large gamme de sévérité, allant de la sécheresse oculaire à la perforation sclérale. Les signes importants pour comprendre le PUK sont présentés ci-dessous.
Kératoconjonctivite sèche : fréquemment associée en tant que syndrome de Sjögren secondaire.
Épisclérite : inflammation des plexus vasculaires superficiels, comme le plexus de la capsule de Tenon. Douleur légère, vision normale.
Sclérite (diffuse ou nodulaire) : congestion des vaisseaux profonds avec douleur oculaire. Inflammation granulomateuse et vascularite prédominantes.
Sclérite nécrosante : zone ischémique jaune-blanc centrale. Pronostic sombre ; sans traitement précoce, cécité et difficulté à conserver le globe oculaire.
Scléromalacie perforante : survient chez les patients traités à long terme pour PR. Amincissement scléral lent sans hyperhémie ni douleur autour de la zone amincie, avec exposition de l’uvée.
Ulcère cornéen périphérique : fréquent dans la PR maligne, avec amincissement rapide et perforation.
QDans quel type de polyarthrite rhumatoïde les ulcères cornéens sont-ils les plus fréquents ?
A
L’ulcère cornéen périphérique et la sclérite nécrosante surviennent facilement dans la PR maligne compliquée de vascularite. La PR avec manifestations extra-articulaires telles que sclérite, pleurésie, pneumonie interstitielle, péricardite, mononévrite multiple et ulcères digitaux est incluse dans ce type, avec un pronostic sombre tant oculaire que systémique. Le contrôle de l’activité de la PR est la base du traitement du PUK.
Le PUK est secondaire à diverses maladies systémiques. On les classe en maladies auto-immunes, infectieuses et autres.
Auto-immunes (les plus fréquentes)
Polyarthrite rhumatoïde (PR) : représente 34 à 42 % des patients atteints de PUK, la cause la plus fréquente 2). Le risque est particulièrement élevé dans la PR maligne compliquée de vascularite, pouvant conduire rapidement à une perforation.
Vascularites associées aux ANCA : la granulomatose avec polyangéite (GPA, anciennement granulomatose de Wegener) provoque une sclérite et un ulcère cornéen périphérique résistants au traitement standard. Le taux de positivité du PR3-ANCA est d’environ 80 %.
Lupus érythémateux disséminé (LED) : survient fréquemment chez les femmes dans la vingtaine et la trentaine.
Polychondrite chronique atrophiante : peut s’accompagner de sclérite ou d’uvéite.
Maladies inflammatoires chroniques de l’intestin (MICI) : 2 à 5 % des patients atteints de MICI présentent des symptômes oculaires tels qu’épisclérite, sclérite ou uvéite9).
Infectieux et autres
Vascularite des gros vaisseaux : des cas de PUK associés à l’artérite à cellules géantes (ACG) ont été rapportés. La TEP-FDG a été utile pour le diagnostic2).
Hidrosadénite suppurée (HS) : cause rare, mais un partage de la voie Th17 est suggéré3)7). L’inflammation oculaire la plus fréquente chez les patients atteints de HS est l’uvéite antérieure, et la PUK est rare.
Rosacée granulomateuse : l’inflammation chronique et l’activation de Th17 sont liées, et des cas de PUK ont été rapportés1).
Inhibiteurs de points de contrôle immunitaires : une PUK bilatérale a été rapportée comme événement indésirable lié à l’immunité (irAE) de la thérapie combinée ipilimumab/nivolumab5).
Infectieux : un cas de canaliculite lacrymale à Citrobacter koseri a provoqué une PUK6). Des cas de PUK associés à la tuberculose ont également été rapportés4).
QQuelles maladies systémiques provoquent la PUK ?
A
La cause la plus fréquente est la polyarthrite rhumatoïde, représentant environ un tiers des patients atteints de PUK. Les autres causes principales sont les vascularites associées aux ANCA (comme la granulomatose avec polyangéite), la polyartérite noueuse, le lupus érythémateux disséminé, la polychondrite chronique atrophiante, les maladies inflammatoires chroniques de l’intestin et d’autres maladies du collagène/auto-immunes. Plus rarement, elle peut survenir dans le cadre d’une hidrosadénite suppurée, d’une rosacée granulomateuse, ou comme événement indésirable lié à l’immunité dû aux inhibiteurs de points de contrôle immunitaires. Les causes infectieuses rapportées incluent la canaliculite bactérienne et la tuberculose.
Examen à la lampe à fente : évaluer l’étendue et la profondeur de l’infiltrat, de l’ulcération et de l’amincissement près du bord de la cornée. Vérifier la présence de défauts épithéliaux à l’aide d’une coloration à la fluorescéine.
Différenciation entre sclérite et hyperhémie conjonctivale/épisclérale : L’instillation d’épinéphrine diluée à 1/1000 fait disparaître l’hyperhémie conjonctivale et limbique, mais pas la dilatation vasculaire due à la sclérite.
Échographie oculaire (mode B) : Utile pour évaluer la sclérite postérieure. On observe un épaississement scléral, des nodules scléraux, un décollement de la capsule de Tenon par rapport à la sclère, et des signes de myosite extra-oculaire.
Frottis et culture cornéens : Réalisés pour exclure une cause infectieuse. En cas de forte suspicion d’infection, une biopsie de la lésion peut être envisagée.
OCT du segment antérieur (AS-OCT) : Utile en cas d’opacité cornéenne sévère rendant difficile l’évaluation de la profondeur à la lampe à fente. L’AS-OCT permet de classer l’ulcère cornéen périphérique (PUK) en trois stades :
Stade aigu : Disparition de l’épithélium cornéen, destruction du stroma antérieur avec réflexion hétérogène, et amincissement cornéen dans la zone lésée.
Stade de cicatrisation : Épithélium irrégulier en régénération présentant une faible réflexion, et homogénéisation de la réflexion stromale.
Stade de guérison : Formation d’une ligne de démarcation nette entre l’épithélium à faible réflexion et le stroma à forte réflexion, avec stabilisation de l’épaississement et de l’amincissement cornéens cicatriciels.
Pour identifier la maladie sous-jacente, les examens suivants sont réalisés de manière systématique. Pour le diagnostic de la polyarthrite rhumatoïde, une augmentation de la VS, une élévation de la CRP et une positivité du facteur rhumatoïde (FR) sont utiles (le FR est négatif dans environ 1/4 des cas). La MMP-3 est un marqueur utile de la prolifération synoviale et est également utilisée pour évaluer l’activité de la maladie.
D’autres examens tels que la numération formule sanguine, la fonction rénale, l’analyse d’urine, les tests de dépistage des hépatites B et C, la radiographie des articulations sacro-iliaques et la tomodensitométrie des sinus sont ajoutés si nécessaire. En cas de suspicion de vascularite des gros vaisseaux, la TEP-FDG est utile, avec une sensibilité de 90 % et une spécificité de 98 % rapportées 2).
Zone claire entre l’ulcère et le limbe. Allergie de type III au staphylocoque.
L’ulcère cornéen catarrhal est une infiltration et ulcération aseptiques de la périphérie cornéenne dues à une réaction allergique de type III aux toxines extracellulaires du staphylocoque. Il survient préférentiellement aux positions 2, 4, 8 et 10 heures, et se distingue de l’ulcère périphérique ulcéreux (PUK) par la présence d’une zone claire de 1 à 2 mm entre l’ulcère et le limbe. L’ulcère de Mooren débute soudainement dans un ou les deux yeux, se présentant comme un ulcère arqué le long du limbe avec un bord creusé abrupt, mais sans zone claire entre l’ulcère et le limbe.
Le traitement du PUK vise à contrôler l’inflammation, prévenir la surinfection, favoriser la cicatrisation de l’ulcère et prévenir la perforation. En cas d’association à une collagénose, le traitement de la maladie sous-jacente est prioritaire et doit être mené en collaboration avec un interniste ou un rhumatologue. Le PUK ne peut être contrôlé si la polyarthrite rhumatoïde n’est pas bien maîtrisée. Un traitement local seul peut permettre un contrôle inflammatoire à court terme, mais si l’activité systémique est élevée, le risque de récidive et de perforation persiste. Par conséquent, le plan de traitement doit être établi conjointement par l’ophtalmologiste et l’interniste.
Larmes artificielles et bouchons lacrymaux : utilisés pour traiter la sécheresse oculaire et favoriser l’épithélialisation. Les préparations sans conservateur sont recommandées. Des collyres à l’acide hyaluronique (Hyalein®) et au rébamipide (Mucosta®) peuvent être associés.
Collyres antibiotiques : utilisés pour prévenir la surinfection.
Collyre à la cyclosporine à 0,05 % : utilisé pour supprimer l’inflammation locale. Comme les collyres immunosuppresseurs commerciaux ne sont pas indiqués, on utilise une préparation maison à 0,05 %. Pour l’ulcère de Mooren, des collyres au tacrolimus ont également été rapportés.
Injection sous-conjonctivale de dexaméthasone : injection de 0,4 mL de solution injectable Décadron® dans la zone d’hyperémie conjonctivale près de l’ulcère.
Collyres stéroïdiens locaux : une extrême prudence est nécessaire car ils peuvent favoriser la kératolyse. En cas de sclérite légère associée, on peut débuter par un collyre à la bétaméthasone à 0,1 % 4 à 6 fois par jour.
Doxycycline orale : utilisée en association pour son effet inhibiteur de la collagénase. La vitamine C peut également être associée pour favoriser la reconstruction du collagène.
Inhibiteur de la COX-2 (célécoxib) : en cas de sclérite légère, 100 mg de célécoxib deux fois par jour par voie orale. Il est très efficace contre la douleur et le contrôle de l’inflammation. En l’absence de contre-indications telles que l’asthme, il doit être associé activement dès le début.
Colle cyanoacrylate : appliquée au fond de l’ulcère en cas de perforation imminente 5).
Le traitement immunosuppresseur systémique est au cœur du traitement.
Prednisolone orale : Dans la PUK, débuter à 1-1,5 mg/kg/jour. En traitement initial de la sclérite associée, utiliser 0,5-1 mg/kg/jour.
Traitement par bolus de corticoïdes : En cas de sclérite nécrosante ou de PUK rapidement progressive, administrer un bolus de méthylprednisolone à 1 g/jour pendant 3 jours.
Immunosuppresseurs (épargneurs de corticoïdes) : Introduire précocement en cas de menace de perforation, de résistance aux stéroïdes ou d’association à la PR.
Méthotrexate (MTX) : Traitement de première intention pour la PUK et la sclérite associées à la polyarthrite rhumatoïde. Également utilisé précocement comme DMARD pour la PR elle-même.
Cyclophosphamide : Choisi pour la PUK et la sclérite nécrosante associées au lupus érythémateux disséminé, aux vascularites à ANCA, à la périartérite noueuse et autres vascularites systémiques.
Azathioprine : Indiqué dans la PR réfractaire et les vascularites, mais son efficacité dans la sclérite serait inférieure à celle du MTX et du cyclophosphamide ; la sélection nécessite une attention particulière.
Ciclosporine : Depuis 2013, l’indication a été élargie pour les uvéites non infectieuses et les scléro-uvéites. Débuter à 2-3 mg/kg/jour et ajuster pour que le taux résiduel ne dépasse pas 150 ng/mL. Effet secondaire : insuffisance rénale, nécessitant des analyses sanguines régulières. Contre-indiqué dans la sclérite associée à la maladie de Behçet neurologique.
Biothérapies : Introduites dans les cas réfractaires résistants aux immunosuppresseurs.
Anti-TNF-α : L’infliximab (Remicade®) et l’adalimumab (Humira®) sont utilisés. En tant que DMARD pour la PR, ils montrent une grande efficacité pour inhiber la destruction osseuse et sont également efficaces dans la scléro-uvéite.
Rituximab : Anticorps anti-CD20. Utilisé dans la PUK réfractaire et les vascularites.
Attention à l’étanercept : Des réactions paradoxales induisant une inflammation oculaire, y compris une sclérite, ont été rapportées avec cet anti-TNF ; il n’est pas recommandé dans la sclérite.
L’adalimumab s’est avéré efficace dans la PUK associée à l’hidradénite suppurée, selon un rapport7).
Traitement DMARD de la polyarthrite rhumatoïde elle-même : Utiliser précocement et activement les antirhumatismaux modificateurs de la maladie pour prévenir la progression de la destruction articulaire. Les immunosuppresseurs comme le méthotrexate et les DMARDs biologiques comme les anti-TNF-α sont centraux. Ces dernières années, l’utilisation précoce de biothérapies a permis de freiner la destruction osseuse et d’améliorer considérablement la qualité de vie à long terme. Pour le soulagement des symptômes articulaires, les AINS et les corticoïdes oraux sont utilisés à court terme.
Prévention des infections : avant et pendant l’utilisation d’immunosuppresseurs ou de biothérapies, un bilan systémique est nécessaire, incluant le dépistage de la réactivation du virus de l’hépatite B et des infections latentes comme la tuberculose.
QPeut-on utiliser des collyres stéroïdiens pour traiter la PUK ?
A
Les collyres stéroïdiens locaux peuvent favoriser la fonte cornéenne (kératolyse), ils doivent donc être utilisés avec prudence dans le traitement de la PUK. Le traitement principal est l’immunosuppression systémique, avec de la prednisolone orale à 1-1,5 mg/kg/jour comme base, associée au méthotrexate en cas de polyarthrite rhumatoïde, ou au cyclophosphamide en cas de vascularite. En traitement local, les collyres de ciclosporine et la doxycycline orale sont recommandés.
La perforation cornéenne ou son imminence est une indication d’intervention chirurgicale.
Conjonctivectomie (opération de Brown) : efficace contre l’ulcère de Mooren. Réséquer la conjonctive hyperémique sur 3-4 mm de large à partir du limbe, jusqu’à 2 heures de part et d’autre de l’ulcère. Également applicable aux ulcères périphériques associés aux collagénoses.
Kératoplastie épithéliale : après conjonctivectomie, si la sclère est exposée, la conjonctive pathologique peut se réétendre. On suture donc 2-3 greffons épithéliaux cornéens minces prélevés sur une cornée de donneur, en forme de digue, au niveau du limbe. Un port de lentille de contact est nécessaire en postopératoire.
Kératoplastie superficielle : réalisée lorsque la maladie est active et que la perforation est inévitable. Utiliser une cornée conservée, gratter suffisamment le tissu prolifératif au fond de l’ulcère avec un couteau golf, puis transplanter. Peut être combinée avec l’opération de Brown ou la kératoplastie épithéliale.
Kératoplastie lamellaire : des cas de perforation de PUK liée aux inhibiteurs de points de contrôle ont été traités par mini-kératoplastie lamellaire5).
Greffe de fronde de la capsule de Tenon : pour les cas avancés de PUK avec cornée en sablier, une greffe circulaire modifiée de la capsule de Tenon a été rapportée4). Avantage : tissu autologue, pas de rejet.
Greffe de patch : en cas de scléromalacie perforante, une réparation chirurgicale de la sclère par greffe de patch peut prévenir l’atrophie du globe.
En postopératoire, il est nécessaire de poursuivre l’administration locale et systémique de stéroïdes ou d’immunosuppresseurs pour prévenir le rejet. Dans les cas de perforation sur fond de sclérite, l’amincissement scléral est étendu et la maladie persiste souvent après l’opération, d’où la nécessité d’une association avec un traitement immunosuppresseur systémique.
Des cas d’injection sous-conjonctivale d’implant de dexaméthasone (Ozurdex) ont été rapportés comme efficaces dans la PUK. Chez les patients âgés ne pouvant tolérer un traitement immunosuppresseur systémique, trois injections ont permis de contrôler l’inflammation pendant 11 mois sans fonte sclérale ni augmentation de la pression intraoculaire8).
La pathogénie de la PUK implique à la fois l’immunité humorale et cellulaire. Dans la polyarthrite rhumatoïde et les vascularites, une surproduction de cytokines inflammatoires se produit dans la synoviale articulaire et la paroi vasculaire, créant un terrain de réaction allergique de type III dans tout l’organisme. Les complexes immuns produits par les anticorps auto-réactifs se déposent dans les vaisseaux du limbe cornéen et de la périphérie, activant la voie classique du complément. L’activation du complément induit la chimiotaxie des neutrophiles et des macrophages, qui libèrent des collagénases et des protéases.
Les cytokines pro-inflammatoires (TNF-α, IL-6, etc.) stimulent les kératocytes pour produire des métalloprotéinases matricielles (MMP)1). Les MMP dégradent le collagène de la périphérie cornéenne, entraînant un amincissement cornéen et la progression de l’ulcère. Dans la polyarthrite rhumatoïde, l’infiltration lymphocytaire de la synoviale, l’angiogenèse et la prolifération synoviale se produisent, et les cytokines inflammatoires provoquent la destruction tissulaire via la mort des chondrocytes et l’activation des ostéoclastes ; un mécanisme similaire est supposé agir dans la périphérie cornéenne.
Récemment, le rôle des cellules Th17 et de l’IL-17 a attiré l’attention1). Les cytokines TGF-β1, IL-6 et IL-21 favorisent la différenciation Th17, et l’IL-1 stimule davantage cette différenciation. L’IL-17 sécrétée par les Th17 augmente la production de MMP, favorisant la dégradation du stroma cornéen1). Il a été rapporté que la suppression des Th17 réduit les complications oculaires de la PUK1).
Dans la rosacée granulomateuse, l’IL-37 est libérée de l’épiderme, activant les mastocytes qui libèrent des protéases telles que la chymase, la tryptase et les MMP1). Ce microenvironnement inflammatoire favorise la polarisation Th17, conduisant au développement de la PUK1).
En cas d’ulcère cornéen périphérique ou de sclérite nécrosante, le pronostic visuel est souvent mauvais. La sclérite nécrosante survient principalement dans la soixantaine, est bilatérale dans environ 60 % des cas, et sans traitement précoce approprié, elle peut conduire à la cécité ou à la perte du globe oculaire. Dans la polyarthrite rhumatoïde maligne, les complications vasculaires systémiques (pulmonaires, cardiaques, rénales) affectent non seulement la fonction visuelle mais aussi le pronostic vital. La scléromalacie perforante survient typiquement chez les patients traités à long terme pour la PR, et au moment où les signes typiques apparaissent, la fenêtre thérapeutique est souvent déjà dépassée. Un diagnostic précoce et une intervention thérapeutique rapide sont essentiels pour améliorer le pronostic.
Raisons pour lesquelles la périphérie cornéenne est un site de prédilection
Le limbe cornéen est la frontière entre l’épithélium cornéen et l’épithélium conjonctival, une zone dense en vaisseaux sanguins, système immunitaire et nerfs. Le limbe contient de nombreuses cellules de Langerhans impliquées dans la présentation antigénique. Alors que la cornée centrale est avasculaire, la région périphérique reçoit facilement des complexes immuns et des cellules inflammatoires du réseau vasculaire limbique. En raison de cette anatomie, dans les ulcères périphériques associés aux maladies du collagène, les complexes immuns se déposent dans le limbe et la périphérie, déclenchant une réaction allergique de type III, et les enzymes de dégradation de la matrice extracellulaire libérées par les cellules immunitaires infiltrées contribuent à la formation de l’ulcère.
QPourquoi les lésions surviennent-elles à la périphérie de la cornée ?
A
Le limbe cornéen est une zone dense en vaisseaux sanguins, système immunitaire et nerfs, et contient de nombreuses cellules de Langerhans. Alors que la cornée centrale est avasculaire, la région périphérique reçoit facilement des complexes immuns et des cellules inflammatoires du réseau vasculaire limbique. Dans les maladies auto-immunes, les complexes immuns se déposent dans les vaisseaux limbiques, activent le système du complément, et les neutrophiles et macrophages s’accumulent pour détruire le stroma cornéen, ce qui explique pourquoi les lésions surviennent préférentiellement à la périphérie.
Avec la généralisation des inhibiteurs de points de contrôle immunitaires (ICI), des PUK en tant qu’irAE ont été rapportés. Dans le traitement combiné par ipilimumab (anticorps anti-CTLA-4) et nivolumab (anticorps anti-PD-1), l’incidence des irAE dépasse 90 % 5). Les PUK liés aux ICI peuvent être résistants aux collyres de stéroïdes à haute dose et à la cyclosporine, et des cas nécessitant une greffe de cornée lamellaire ont été rapportés 5). L’équilibre entre la poursuite du traitement antitumoral et la gestion des irAE est un défi, et la collaboration avec l’oncologie est essentielle.
Pour les patients qui ne tolèrent pas un traitement immunosuppresseur systémique, l’injection sous-conjonctivale d’un implant de dexaméthasone (Ozurdex) a été tentée 8). Des administrations répétées ont permis un contrôle inflammatoire à long terme sans scléromalacie ni augmentation de la pression intraoculaire8). Ce traitement local évitant les effets secondaires systémiques mérite d’être évalué à l’avenir.
Étant donné que la voie Th17/IL-17 est profondément impliquée dans la pathologie de la PUK, les thérapies ciblant Th17 suscitent l’intérêt. Dans un cas de PUK associée à une rosacée granulomateuse ayant répondu à l’isotrétinoïne, il a été suggéré que l’effet inhibiteur de l’isotrétinoïne sur Th17 et son effet promoteur sur les lymphocytes T régulateurs pourraient être impliqués 1).
Une greffe de fronde de la capsule de Tenon modifiée a été rapportée pour les PUK avancées 4). Comme il s’agit de tissu autologue, il n’y a pas de rejet et l’intervention est possible même lorsque la cornée de donneur est difficile à obtenir 4).
Hsiao FC, Meir YJ, Hsiao CH, et al. Peripheral ulcerative keratitis in a patient with granulomatous rosacea. Taiwan J Ophthalmol. 2023;13(1):80-83. doi:10.4103/tjo.tjo-d-22-00079.
Uchida S, Kaji Y, Ui M, Kawashima H, Usui T, Ohira Y. Peripheral Ulcerative Keratitis Associated With Large Vessel Vasculitis. Cureus. 2021;13(6):e15767. doi:10.7759/cureus.15767. PMID:34290940; PMCID:PMC8290306.
Dallalzadeh LO, Ang MJ, Beazer AP, Spencer DB, Afshari NA. Peripheral ulcerative keratitis secondary to severe hidradenitis suppurativa. American journal of ophthalmology case reports. 2022;25:101403. doi:10.1016/j.ajoc.2022.101403. PMID:35198822; PMCID:PMC8844392.
Anitha V, Ghorpade A, Ravindran M. A modified Tenons sling annular graft for advanced peripheral ulcerative keratitis with an hourglass cornea. Indian J Ophthalmol. 2022;70(2):655-657. doi:10.4103/ijo.IJO_2035_21. PMID:35086257; PMCID:PMC9023998.
Aschauer J, Donner R, Lammer J, Schmidinger G. Bilateral corneal perforation in Ipilimumab/Nivolumab-associated peripheral ulcerative keratitis. Am J Ophthalmol Case Rep. 2022;28:101686. doi:10.1016/j.ajoc.2022.101686.
Acharya I, Jalloh MI, Trevisan CD, Haas CJ. Hidradenitis Suppurativa and Peripheral Ulcerative Keratitis. Journal of community hospital internal medicine perspectives. 2024;14(6):89-93. doi:10.55729/2000-9666.1408. PMID:39839173; PMCID:PMC11745191.
Ghoraba HH, Or C, Ahluwalia A, Mekonnen B, Yasar C, Yavari N, et al. Subconjunctival dexamethasone implant (Ozurdex) for peripheral ulcerative keratitis. American journal of ophthalmology case reports. 2025;39:102379. doi:10.1016/j.ajoc.2025.102379. PMID:40686761; PMCID:PMC12274858.
Campagne O, Vinet E, Engel L, et al. Ocular manifestations of inflammatory bowel disease. Ocul Surf. 2023;29:326-339.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.