Le mélanome malin primitif de la conjonctive est une tumeur maligne provenant des mélanocytes conjonctivaux. Il est fréquent en Occident mais rare au Japon. La mélanose acquise primaire (PAM) en est souvent le précurseur.
L’incidence mondiale est de 0,3 à 0,8 par million d’habitants par an, la plus élevée en Scandinavie et en Amérique du Nord. Elle a augmenté au cours des 50 dernières années. On estime environ 130 nouveaux cas par an aux États-Unis et 320 en Europe 8). Le taux d’incidence ajusté sur l’âge chez les Asiatiques est faible (0,15 par million par an), et il est plus fréquent chez les Blancs (91,2 %) 1, 8).
L’âge moyen d’apparition est de 55 à 65 ans, et l’apparition avant 20 ans est extrêmement rare 1, 4, 5, 6). Le taux de survie spécifique à 5 ans est d’environ 82,9 %, et à 10 ans de 69,3 % 8).
Répartition par origine :
Dérivé de PAM (mélanose acquise primaire) : environ 60 à 75 % (le plus fréquent)
De novo : environ 19 %
Dérivé d’un naevus conjonctival : 7 à 20 %
Le mélanome conjonctival est le seul mélanome des muqueuses pour lequel une association avec l’exposition aux UV est suggérée, car la conjonctive bulbaire est directement exposée aux rayons ultraviolets 4).
QÀ quel point le mélanome malin de la conjonctive est-il rare ?
A
L’incidence du mélanome malin de la conjonctive est rare, de 0,3 à 0,8 par million de personnes par an, mais elle a augmenté au cours des 50 dernières années. Les Asiatiques ont un risque plus faible que les Caucasiens, avec 0,15 par million de personnes par an. Les cas pédiatriques ne représentent que 1 % de tous les mélanomes conjonctivaux, et l’apparition avant 20 ans est extrêmement rare.
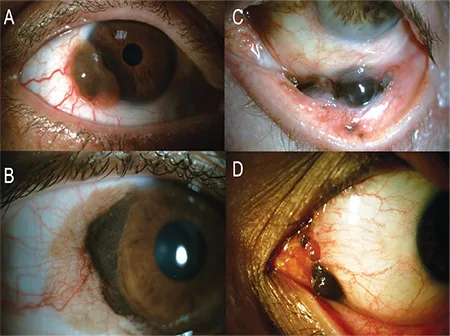
Koç İ, Kiratlı H. Current Management of Conjunctival Melanoma Part 1: Clinical Features, Diagnosis and Histopathology. Turk J Ophthalmol. 2020;50(5):293-303. Figure 1. PMCID: PMC7610047. License: CC BY.
A) Mélanome conjonctival limbique pigmenté surélevé avec des vaisseaux nourriciers abondants, B) Grand mélanome limbique survenant sur un fond de PAM diffus, C) Mélanome localisé dans le fornix, D) Photographie du segment antérieur montrant un mélanome s’étendant à la caroncule et au pli semi-lunaire. Correspond aux sites et formes cliniques du mélanome malin de la conjonctive traités dans la section « 2. Principaux symptômes et signes cliniques ».
Cliniquement, il s’agit d’une lésion brun-noir surélevée de la conjonctive bulbaire ou palpébrale, avec une vascularisation abondante en direction de la tumeur.
Mélanome pigmenté (70 %)
Sites de prédilection : environ 90 % surviennent sur la conjonctive bulbaire, et 63 % sont localisés dans le quadrant temporal 4, 6).
Aspect : lésion surélevée de couleur brun noir à brun foncé. Présence de vaisseaux nourriciers abondants (feeder vessels) se dirigeant vers la tumeur.
Relation avec la sclère : une adhérence à la sclère peut être observée. La distance moyenne entre la tumeur et le limbe cornéen est de 2 mm, et 61 % atteignent le limbe2).
Mélanome amélanotique (30 %)
Aspect : masse de couleur rose à rouge, contenant peu ou pas de pigment.
Risque de diagnostic erroné : facilement confondu avec un carcinome épidermoïde ou d’autres masses conjonctivales rouges 3).
Pronostic : les mélanomes hypopigmentés ou amélanotiques peuvent être associés à un pronostic défavorable.
Microscopie ultrasonore biomicroscopique (UBM) : mesure de l’épaisseur de la tumeur et vérification de l’infiltration sclérale 3)
QExiste-t-il un mélanome conjonctival sans pigment ?
A
Oui. Environ 30 % des mélanomes conjonctivaux sont amélanotiques et se présentent comme une masse rose à rouge. Le mélanome amélanotique ressemble à un carcinome épidermoïde et est souvent mal diagnostiqué, ce qui peut entraîner un retard de diagnostic. Une biopsie avec diagnostic pathologique est indispensable pour toute masse conjonctivale suspecte.
Les facteurs suivants sont associés à un risque accru de métastase et de décès1, 4, 5, 6, 8).
Épaisseur tumorale > 2 mm : HR 1,20 par augmentation de 1 mm
Invasion profonde : HR 2,35
Emboles lymphatiques : HR 7,49
Ulcération : HR 7,01
Tumeur T3 : HR 17,44
Mélanome nodulaire : RR de métastase 6,00–8,44, RR de décès 25,49–35,49
Site de la lésion : Fornix et conjonctive palpébrale > conjonctive bulbaire. Les lésions de la caroncule ont le plus mauvais pronostic avec une mortalité à 3 ans de 50 %
Invasion orbitaire
Résection incomplète : Environ 49,3 % de récidive en cas de résection incomplète
La voie principale de métastase est lymphatique. Les lésions conjonctivales temporales métastasent souvent aux ganglions lymphatiques pré-auriculaires, tandis que les lésions nasales métastasent aux ganglions sous-maxillaires. Les métastases à distance (hématogènes) surviennent dans le cerveau, les poumons, le foie, la peau et les glandes surrénales6, 1). Les métastases ganglionnaires sont observées chez 15 à 41 % des patients dans les 2,3 ans suivant le diagnostic, et les métastases systémiques surviennent chez 9 à 25 % dans les 3 ans. Même en l’absence de métastases ganglionnaires détectées, des métastases viscérales hématogènes à distance surviennent dans 38 % des cas6).
QQuel est le facteur qui influence le plus le pronostic ?
A
Les principaux facteurs de mauvais pronostic sont l’épaisseur tumorale (notamment > 2 mm), le site de la lésion (la caroncule lacrymale étant le plus défavorable), la classification AJCC, la morphologie nodulaire, l’invasion lymphovasculaire et l’invasion orbitaire. La complétude de la résection a également un impact majeur : environ 49,3 % des cas de résection incomplète récidivent. Les cas avec ganglion sentinelle positif présentent un risque accru de décès.
Le diagnostic définitif nécessite une biopsie. Histologiquement, on observe une prolifération de cellules à grand rapport nucléo-cytoplasmique, contenant de la mélanine et présentant des nucléoles proéminents. En cas de mélanine abondante, une décoloration de l’échantillon est nécessaire.
L’immunohistochimie est indispensable pour le diagnostic définitif et le diagnostic différentiel.
La résection chirurgicale de la tumeur incluant les tissus environnants est la base du traitement. L’administration locale per- et postopératoire de MMC ou d’interféron alpha-2b est également efficace. En cas de tumeur étendue avec invasion sous-conjonctivale marquée, une exentération orbitaire peut être nécessaire.
Traitement chirurgical
Marge de résection : Résection complète de la tumeur avec une marge de sécurité de 3 à 5 mm4, 8)
Technique sans contact : Éviter tout contact direct avec la tumeur à l’aide d’instruments pour prévenir la dissémination des cellules tumorales4, 8)
Technique sèche : Résection sans utilisation de solution de perfusion (BSS)
Cas d’infiltration cornéenne : Décollement épithélial cornéen à l’alcool + résection en bloc au couteau hockey2)
Exentération orbitaire : Indiquée dans les cas étendus ou récidivants (stade avancé)
Énucléation : Indiquée en cas d’extension intraoculaire
Traitement adjuvant
Cryothérapie : Appliquée sur la base et les bords de la résection selon la technique de double congélation. Soulever la conjonctive pour éviter les lésions sclérales3)
Chimiothérapie locale à la MMC :
Peropératoire : Application de micro-éponge de MMC à 0,02 % pendant 180 secondes2)
Postopératoire : Gouttes de MMC à 0,02 % 4 fois/jour × 7 jours × 3 cycles2)
IFNα-2b : Alternative à la MMC sans lésion des cellules souches limbiques3)
Radiothérapie : curiethérapie ou radiothérapie externe3)
Biopsie du ganglion sentinelle : envisagée pour une épaisseur tumorale > 2 mm7)
Pour la reconstruction conjonctivale après exérèse large, on utilise les techniques suivantes4, 5) :
Greffe de membrane amniotique : possède des effets anti-inflammatoires, anti-fibrotiques et anti-angiogéniques
Greffe de muqueuse buccale
Greffe conjonctivale controlatérale
QQu'est-ce que la technique « no-touch » ?
A
Il s’agit d’une technique chirurgicale visant à prévenir la dissémination des cellules tumorales (récidive locale ou métastases) en évitant tout contact direct avec la tumeur à l’aide d’instruments et en réalisant l’excision dans un environnement sec, sans utiliser de solution de perfusion (BSS). Une marge de sécurité de 3 à 5 mm est assurée pour retirer la tumeur en un seul bloc, réduisant ainsi le risque de récidive due à une résection incomplète. Cette technique diffère fondamentalement de l’excision standard car elle minimise la dissémination des cellules tumorales sur la conjonctive.
Le mélanome conjonctival est génétiquement similaire au mélanome cutané plutôt qu’au mélanome uvéal. Les principales mutations driver liées aux UV (transition C>T) sont BRAF, NF1 et RAS. Les marqueurs du mélanome uvéal (BAP1, GNAQ, GNA11, SF3B1) sont négatifs dans le mélanome conjonctival1).
La mutation du promoteur TERT (c.-124C>T) affecte la télomérase transcriptase inverse et a été associée au mélanome conjonctival métastatique1, 8, 6). Elle est également détectée dans les atypies modérées à sévères du PAM (environ 8 %), suggérant une nature de mélanome in situ6). Une association avec une charge mutationnelle tumorale élevée a également été démontrée4). Des mutations de TERT sont observées dans 32 à 64 % des mélanomes conjonctivaux, et leur lien avec le pronostic est étudié8).
Une expression élevée de PD-L1 et la présence d’un sous-type transcriptionnel riche en gènes liés au système immunitaire ont été confirmées, ce qui constitue la base théorique de l’utilisation des inhibiteurs de points de contrôle immunitaires. Les données sur les inhibiteurs de BRAF, de MEK et de PD-L1 sont prometteuses mais encore limitées à ce jour.
Chou et al. (2023) ont analysé le profil moléculaire d’un cas de mélanome conjonctival T3c chez un homme de 94 ans. Ils ont identifié une mutation NF1 et une mutation du promoteur TERT (c.-124C>T, VAF 31,4 %), tandis que BRAF, NRAS et cKIT étaient tous négatifs. La combinaison d’une mutation NF1 et de l’absence de mutation NRAS est considérée comme un facteur contribuant à l’évolution relativement favorable sans métastase1).
Les inhibiteurs de PD-1 (pembrolizumab, nivolumab) et de CTLA-4 (ipilimumab) ont été testés dans le mélanome conjonctival métastatique et localement avancé4, 8).
Un rapport représentatif est celui de Sagiv et al. (2018) qui ont traité 5 patients par inhibiteur de PD-1 (pembrolizumab ou nivolumab) et ont rapporté des réponses complètes chez certains8). Un essai de phase 2 associant axitinib et nivolumab (chez des patients atteints de mélanome muqueux avancé ou métastatique non traité) est en cours.
Le schéma thérapeutique systémique standard pour le mélanome conjonctival métastatique n’est pas encore établi, et une décision prudente au cas par cas est nécessaire.
Thérapie ciblée (inhibiteurs de BRAF, inhibiteurs de MEK)
Pour les cas avec mutation BRAF, l’inhibiteur de BRAF seul (vemurafenib) ou la combinaison d’inhibiteurs BRAF/MEK (dabrafenib + trametinib, encorafenib + binimetinib) sont en cours d’essai 4, 8).
Ces médicaments ont montré une réduction tumorale locale dans certains cas, mais la plupart des rapports concernent des cas uniques ou un petit nombre de cas, et les résultats à long terme sont limités. La possibilité d’un effet synergique par ciblage simultané des voies MAPK et AKT est également à l’étude 3).
Progrès dans les techniques de diagnostic et la classification moléculaire
L’utilisation de panels immunohistochimiques (PRAME, p16, HMB-45, Ki-67, Cyclin D1) permet de reclassifier les lésions auparavant considérées comme « indéterminées » 5). La standardisation internationale de la classification C-MIL (lésion mélanocytaire intraépithéliale conjonctivale) (OMS 5e édition, 2022) progresse également.
QL'immunothérapie peut-elle être utilisée pour le mélanome conjonctival ?
A
Sur la base des similitudes génétiques avec le mélanome cutané (mutations BRAF, NF1, NRAS, forte expression de PD-L1), les inhibiteurs de PD-1 (pembrolizumab, nivolumab), les inhibiteurs de CTLA-4 et les inhibiteurs de BRAF sont essayés dans les cas métastatiques ou localement avancés. Cependant, aucun essai clinique à grande échelle n’a été mené, les preuves sont actuellement limitées et aucun schéma thérapeutique standard n’est établi. L’accumulation de données à grande échelle par le biais d’études multicentriques internationales est considérée comme urgente.
Chou LT, Lozeau DF, Boyle NS. A rare case of a long-standing, extensive and invasive conjunctival melanoma without systemic metastasis. Am J Ophthalmol Case Rep. 2023; PMC10121375.
Englisch CN, Berger T, Flockerzi F, et al. Conjunctival melanoma with pronounced central corneal invasion: one-year relapse free follow-up. Am J Ophthalmol Case Rep. 2024; PMC11403272.
Menna F, Tschopp M, Meyer P, et al. A case of conjunctival melanoma presenting as a squamous cell carcinoma. Case Rep Ophthalmol. 2024; PMC11509494.
Okongwu CC, Adewara BA, Olaofe OO, Soremekun AI, Ayodele SO, Abdullahi YO, et al. Malignant melanoma of the conjunctiva metastasizing to the submandibular gland: a case report and review of the literature. BMC Ophthalmol. 2025;25(1):130. doi:10.1186/s12886-025-03949-5. PMID:40082862. PMCID:PMC11905526.
Eder A, Milman T, Mudhar HS, et al. Unusual conjunctival melanocytic proliferations: report of five cases and review of the literature. Surv Ophthalmol. 2024; PMC12208716.
Goemaere J, Lauwers N, de Keizer ROB, et al. Bone metastasis in a case of primary acquired melanosis with atypia resulting from conjunctiva melanoma. Am J Ophthalmol Case Rep. 2023;29:101730. doi:10.1016/j.ajoc.2022.101730. PMID:36561878; PMCID:PMC9763362.
Vishnevskia-Dai V, Davidy T, Zloto O. Amelanotic conjunctival melanoma in a child. Am J Ophthalmol Case Rep. 2023; PMC9792290.
Butt K, Hussain R, Coupland SE, Krishna Y. Conjunctival melanoma: a clinical review and update. Cancers. 2023;15(3):922.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.