El síndrome de Aicardi es una enfermedad congénita rara descrita por primera vez en 1967 por el neurólogo francés Jean Aicardi. Se presume que es de herencia dominante ligada al cromosoma X, y casi todos los pacientes son niñas. En los niños, es letal en estado hemicigoto, por lo que solo se han reportado unos pocos casos en niños con cariotipo XXY (síndrome de Klinefelter).
La incidencia se estima en aproximadamente 1:110,000 nacimientos, y el número de personas afectadas en todo el mundo es de alrededor de 4,000 1). Todos los casos son mutaciones de novo, sin transmisión de padres a hijos, y el riesgo de recurrencia para hermanos es menor del 1% 1).
La tríada clásica consta de las siguientes tres características 1):
Espasmos infantiles: inicio alrededor de los 3-4 meses de edad.
Lagunas coriorretinianas (chorioretinal lacunae): Lesiones fundoscópicas redondas bilaterales. Un hallazgo específico de esta enfermedad.
Agenesia del cuerpo calloso (agenesis of the corpus callosum): Ausencia parcial o completa.
El pronóstico es malo. La edad media de supervivencia es de 18 años, y la probabilidad de sobrevivir hasta los 27 años se reporta en 0.62% 1).
Q¿El síndrome de Aicardi ocurre en niños varones?
A
Esta enfermedad ocurre casi exclusivamente en niñas. Se presume que es hereditaria dominante ligada al X, ya que es letal en varones hemicigotos. Sin embargo, existen algunos casos reportados en todo el mundo en niños con cariotipo XXY (síndrome de Klinefelter).
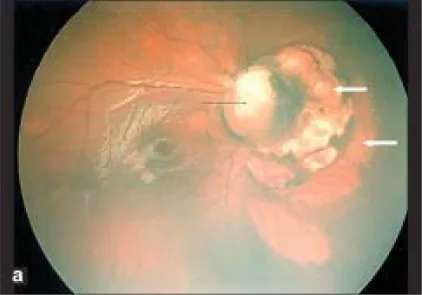
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Foto Retcam del ojo derecho muestra coloboma del disco óptico (flecha negra) y áreas pálidas en forma de cúpula con bordes definidos nasales al disco óptico sugestivas de lagunas coriorretinianas (flechas blancas).
El síntoma inicial de esta enfermedad suele ser el espasmo infantil que aparece alrededor de los 3-4 meses de edad. La epilepsia a menudo se vuelve farmacorresistente y se acompaña de diversos tipos de convulsiones.
Convulsiones epilépticas: Inicialmente se presentan como espasmos infantiles, progresando a farmacorresistencia. En un caso, ocurrieron convulsiones generalizadas 3-4 veces al día (cada una de 20-25 minutos de duración) a los 4 meses de edad 1). En otro caso, se observaron frecuentes convulsiones de parpadeo desde el mes de edad 2).
Retraso psicomotor: Acompañado de discapacidad intelectual grave, a menudo dificulta el movimiento independiente y la adquisición del lenguaje.
Disfunción gastrointestinal: Los síntomas gastrointestinales como el estreñimiento están presentes en más del 90% de los casos 1).
Discapacidad visual: Discapacidad visual debida a lesiones del fondo de ojo, agenesia del cuerpo calloso y malformaciones corticales.
En informes de casos se ha documentado un ejemplo con hemorragia prerretiniana y zona avascular periférica de 360 grados en el ojo derecho, y tejido pediculado (pedículo fibrovascular) con desprendimiento de retina traccional en el ojo izquierdo2). También puede causar deterioro visual cortical (CVI)3).
Agenesia del cuerpo calloso: La ausencia parcial o completa está presente en todos los casos1). También se ha reportado adelgazamiento del cuerpo calloso (disgenesia) como una variante2).
Malformaciones de la corteza cerebral: Polimicrogiria (tipo 2 de Barkovich), heterotopia nodular periventricular y quistes multicamerales se confirman en la RM1).
Hallazgos en el EEG: Patrón característico de ritmos polimórficos de alto voltaje con puntas y ondas lentas multifocales1).
Anomalías esqueléticas: Fusión vertebral torácica (T9–T10) y vértebra en mariposa (T8) se observan en el 40–60% de los casos1).
Q¿Las lagunas coriorretinianas cambian con el tiempo?
A
Pueden progresar. Se ha informado que aparecen nuevas lagunas después de una intervención quirúrgica oftalmológica, o que su tamaño o número aumentan con el tiempo2). Es importante realizar un seguimiento periódico con examen de fondo de ojo.
El síndrome de Aicardi se presume hereditario dominante ligado al X, pero el gen causante no se ha identificado hasta la fecha1). Todos los casos son mutaciones de novo y no se observa aparición familiar. El riesgo de recurrencia para hermanos es menor del 1%, y se recomienda considerar la planificación de futuros hijos tras asesoramiento genético1).
En un informe de caso reciente, se detectó una mutación en el gen TREX1 (c.292_293insA, p.(Cys99Metfs)) en un paciente2). TREX1 se localiza en el cromosoma 3, lo que contradice parcialmente la hipótesis de ligamiento al X, por lo que la ubicación del gen causante sigue siendo debatida.
Patrón de herencia: Dominante ligado al X (presunto). Los varones hemicigotos son letales en el útero.
Naturaleza de la mutación: Todas son mutaciones de novo. No hay herencia (transmisión de padres a hijos).
Riesgo de recurrencia: Menor del 1% para hermanos1).
Dado que el gen causante no se ha identificado, el diagnóstico del síndrome de Aicardi es principalmente clínico. La confirmación de la tríada clásica es fundamental para el diagnóstico1).
Espasmos infantiles (infantile spasms)
Lagunas coriorretinianas (chorioretinal lacunae)
Agenesia del cuerpo calloso (agenesis of the corpus callosum)
Incluso si solo se cumplen dos de las tres características cardinales, el diagnóstico es posible utilizando los criterios diagnósticos ampliados establecidos en 1999.
Los componentes de los criterios diagnósticos ampliados se muestran a continuación.
RMN (cabeza): Confirmación de agenesia del cuerpo calloso. Evaluar polimicrogiria, sustancia gris heterotópica, agrandamiento ventricular lateral, quistes estriatales talámicos bilaterales, hipoplasia del hipocampo, etc.1)2)
EEG (electroencefalograma): Confirmar el patrón de ritmos polimórficos de alto voltaje y descargas punta-onda multifocales.1)
Examen de fondo de ojo: Confirmación de lagunas coriorretinianas. La angiografía fluoresceínica (FA) es útil para evaluar áreas avasculares 2).
Radiografía de columna torácica (AP): Confirmación de anomalías esqueléticas (vértebras fusionadas, vértebras en mariposa) 1).
Colaboración multidisciplinaria: Los departamentos de neurología, oftalmología, ortopedia y genética deben colaborar para el diagnóstico 1).
Q¿Pueden las pruebas genéticas proporcionar un diagnóstico definitivo?
A
Actualmente, no se ha identificado ningún gen causal que permita un diagnóstico definitivo, por lo que las pruebas genéticas por sí solas no pueden proporcionar un diagnóstico definitivo 1). El diagnóstico clínico basado en la combinación de síntomas clínicos, hallazgos de fondo de ojo y hallazgos de imagen es el enfoque principal. Se espera que los avances en el análisis genómico integral identifiquen genes causales en el futuro.
No existe un tratamiento curativo. Los objetivos del tratamiento consisten en tres pilares: control de las convulsiones, manejo de las complicaciones oftálmicas y apoyo al desarrollo mediante rehabilitación.
Manejo de las convulsiones
Fármacos de primera línea: Se utilizan fenitoína, levetiracetam, clobazam, etc. 1).
Casos refractarios: Se intentan cannabidiol (CBD), terapia de dieta cetogénica, callosotomía y estimulación del nervio vago 1).
Intervención oftálmica
Fotocoagulación con láser: Se realiza en la retina avascular periférica para prevenir la progresión de la retinopatía proliferativa 2).
Se presentan casos reportados de intervenciones quirúrgicas para complicaciones oftálmicas.
Ojo
Hallazgos
Tratamiento
Ojo derecho
Hemorragia prerretiniana, zona avascular 360 grados
Fotocoagulación con láser
Ojo izquierdo
Tejido en tallo, TRD
Vitrectomía 23G
Después de la vitrectomía, se confirmó la recuperación del crecimiento de la longitud axial; en el ojo izquierdo, la longitud axial creció de 17.45 mm al mes de edad a 24.41 mm a los 26 meses de edad 2). Además, se han reportado casos donde las lagunas coriorretinianas se hicieron visibles después de la cirugía, contribuyendo al diagnóstico definitivo 2).
Q¿Es posible el tratamiento quirúrgico oftálmico?
A
Sí, es posible. La fotocoagulación con láser para la retina avascular periférica y la vitrectomía de 23G para el desprendimiento de retina traccional pueden ser efectivas 2). Hay informes de crecimiento ocular acelerado después de la cirugía. Sin embargo, el número de casos es pequeño y se requiere un manejo cuidadoso en una instalación especializada.
Se presume que las mutaciones de novo en el cromosoma X son la causa de esta enfermedad. El patrón de inactivación del cromosoma X (lionización) se cree que produce variabilidad fenotípica incluso con la misma mutación. En los varones, la hemicigosis conduce a letalidad en el período embrionario, y solo los varones con cariotipo XXY pueden sobrevivir.
Recientemente, se detectó una mutación en el gen TREX1 (c.292_293insA, p.(Cys99Metfs)) en un caso diagnosticado con esta enfermedad 2). Dado que TREX1 se encuentra en el cromosoma 3, esto puede contradecir la hipótesis ligada al X, lo que sugiere la posibilidad de genes causantes no ligados al X.
Histológicamente, las lagunas coriorretinianas se caracterizan por defectos en el epitelio pigmentario de la retina (EPR) que se extienden desde la coriocapilar hasta la esclerótica desnuda 2). El área del defecto carece de EPR y coriocapilar, y se observa neuroretina indiferenciada displásica.
Se cree que la persistencia del sistema vascular fetal contribuye a la formación de tejido de tallo fibrovascular y retina avascular periférica 2). Estos vasos anormales causan desprendimiento de retina traccional.
La discapacidad visual cortical (CVI) surge como una disminución adquirida de la función visual debido a anomalías estructurales del cerebro como polimicrogiria, heterotopia de sustancia gris y agenesia del cuerpo calloso 3).
Las malformaciones cerebrales (polimicrogiria, agenesia del cuerpo calloso) se basan en una alteración de la migración neuronal durante el período embrionario. Los mecanismos moleculares subyacentes a este trastorno de migración siguen siendo desconocidos, y se espera investigación futura junto con la identificación de genes causantes.
7. Investigación más reciente y perspectivas futuras (Informes en fase de investigación)
El gen causante sigue sin identificarse, pero la detección de mutaciones en el gen TREX1 2) es una nueva pista que sugiere la posible participación de mutaciones no ligadas al cromosoma X. Se espera que la difusión del análisis genómico integral (WES/WGS) acelere la identificación del gen causante. La verificación de las hipótesis ligada al X y no ligada al X es un desafío futuro importante.
Reconocimiento del síndrome de Aicardi variante (atípico)
Se están acumulando informes de casos variantes que muestran solo adelgazamiento (disgenesia) en lugar de agenesia completa del cuerpo calloso 2). Incluso en casos que no cumplen con los tres signos cardinales, la aplicación de un examen detallado del fondo de ojo y criterios diagnósticos ampliados puede mejorar la precisión diagnóstica.
Importancia de la intervención oftalmológica temprana
Kang y colaboradores (2022) informaron que la fotocoagulación con láser y la vitrectomía de 23G realizadas para la vitreorretinopatía bilateral en el síndrome de Aicardi contribuyeron a mantener el crecimiento ocular y proteger la función visual 2). También registraron que las lagunas coriorretinianas se hicieron visibles de nuevo después de la cirugía, lo que indica que la cirugía oftalmológica también puede ayudar a confirmar el diagnóstico.
Existe un reconocimiento creciente de que el cribado temprano del fondo de ojo y la intervención rápida son importantes para proteger la función retiniana 2). El tratamiento con láser en las áreas avasculares periféricas puede prevenir la progresión de la retinopatía proliferativa, y se desea una mayor acumulación de casos.
La investigación sobre la aplicación del cannabidiol (CBD) y la terapia con dieta cetogénica para el control de las convulsiones está avanzando 1). Se espera el establecimiento de nuevas opciones de tratamiento para la epilepsia intratable.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.