
Coloboma del disco óptico
Puntos clave de un vistazo
Sección titulada «Puntos clave de un vistazo»1. ¿Qué es el coloboma del disco óptico?
Sección titulada «1. ¿Qué es el coloboma del disco óptico?»El coloboma del disco óptico es una anomalía congénita caracterizada por un disco óptico anormalmente agrandado con una depresión blanca bien definida. Resulta del cierre incompleto de la fisura del cáliz óptico (fisura embrionaria), que normalmente se cierra en la séptima semana de gestación. Los vasos retinianos no se originan de un solo punto, sino de varios lugares en el borde o dentro de la depresión.
Cuando el cierre incompleto de la fisura del cáliz óptico se limita a la parte posterior (lado del nervio óptico), se produce el coloboma del disco óptico. Si el cierre incompleto es más extenso de anterior a posterior, forma un espectro de coloboma de iris a coroides. Dentro del espectro general de coloboma, el coloboma del disco óptico corresponde al extremo posterior del cierre incompleto y es parte de un espectro continuo desde el coloboma de iris (extremo anterior), pero también existe una forma aislada limitada al disco óptico.
El código ICD-10 es H47.319 (nervio óptico).
Es importante diferenciarlo del síndrome de morning glory. En el síndrome de morning glory, se observa tejido de proliferación glial en el centro del disco y los vasos discurren radialmente. El coloboma del disco óptico se puede diferenciar por la ausencia de proliferación glial, una depresión predominante inferior y vasos que se originan en varios sitios en el borde o dentro de la depresión.
El coloboma del disco óptico presenta una depresión blanca bien definida de predominio inferior, y los vasos se originan en varios sitios en el borde o dentro de la depresión. No se observa tejido de proliferación glial. En el síndrome de morning glory, hay tejido de proliferación glial en el centro del disco y los vasos discurren radialmente desde el margen del disco. Ambas son anomalías congénitas del disco óptico, pero se pueden diferenciar por los hallazgos del fondo de ojo.
2. Síntomas principales y hallazgos clínicos
Sección titulada «2. Síntomas principales y hallazgos clínicos»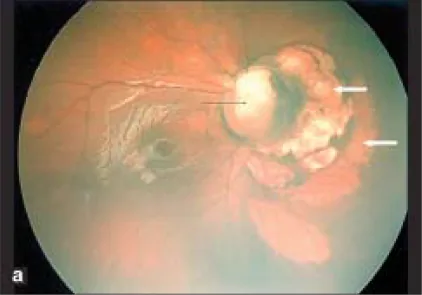
Síntomas subjetivos
Sección titulada «Síntomas subjetivos»La agudeza visual depende de si el haz papilomacular está involucrado en el coloboma y del grado de afectación. Varía desde mejor de 1.0 hasta mala, pero en muchos casos la agudeza visual está reducida incluso sin afectación macular debido a la anomalía del nervio óptico.
- Disminución de la agudeza visual: Depende del grado de afectación del haz papilomacular. En casos de mala visión, puede presentarse estrabismo por desuso.
- Defecto del campo visual: A menudo causa un defecto del campo visual superior correspondiente al coloboma inferior al disco óptico.
- Estrabismo: En casos de mala visión, puede desarrollarse estrabismo por desuso secundario.
Hallazgos del fondo de ojo
Sección titulada «Hallazgos del fondo de ojo»En el fondo de ojo, se producen defectos del disco óptico y de la retina y coroides inferiores al globo ocular, principalmente en la parte inferior. Hay anomalías vasculares; debido a que la arteria central de la retina se ramifica detrás del disco, muchas arterias retinianas parecen emerger del disco. El borde superior del disco a menudo permanece, e incluso cuando todo el disco está deprimido, la depresión es típicamente más fuerte en la parte inferior en comparación con la superior.
El área del disco está deprimida, y el disco está ausente o parcialmente defectuoso; la coroides circundante, el epitelio pigmentario de la retina (EPR) y la esclerótica también son delgados. Inferior a la depresión del área del disco, hay atrofia retino coroidea y aspecto de empedrado debido al cierre incompleto de la fisura embrionaria.
Clasificación
Sección titulada «Clasificación»El coloboma del disco óptico se clasifica de la siguiente manera según la extensión de la afectación.
- Coloboma aislado del disco óptico: Debido al cierre incompleto localizado de la parte posterior de la fisura de la copa óptica.
- Combinado con coloboma retino coroideo: Indica un cierre incompleto más extenso de la fisura de la copa óptica.
- Tipo combinado de coloboma de iris/cuerpo ciliar: Tipo extenso continuo desde el extremo anterior hasta el posterior.
- Coloboma de Fuchs: Tipo leve que muestra una lesión atrófica similar a un cono debajo del disco óptico. La agudeza visual a menudo se conserva relativamente.
Complicaciones
Sección titulada «Complicaciones»Complicaciones oculares
Sección titulada «Complicaciones oculares»- Con frecuencia se asocian coloboma de iris y coloboma coroideo.
- Cuando se asocia coloboma coroideo y el área deprimida es grande, puede presentarse microftalmía.
- Desprendimiento de retina seroso: Puede ocurrir incluso con coloboma del disco óptico solo.
- Desprendimiento de retina regmatógeno: Puede ocurrir secundariamente en casos con coloboma retinoideo-coroideo complejo.
- Hipotensión ocular por filtración transescleral: También se han reportado casos de fuga de humor acuoso desde el defecto escleral7).
3. Causas, epidemiología, factores de riesgo
Sección titulada «3. Causas, epidemiología, factores de riesgo»Epidemiología
Sección titulada «Epidemiología»La prevalencia se reporta en 3–8/100,000. Los casos unilaterales y bilaterales son igualmente comunes, y aunque muchos casos son esporádicos, a menudo hay antecedentes familiares. Se han reportado diversos patrones de herencia, incluyendo autosómico dominante, autosómico recesivo y ligado al X4).
La tasa de diagnóstico genético general del coloboma sigue siendo inferior al 30%5). Muchos casos esporádicos sin mutaciones genéticas identificadas sugieren la participación de factores ambientales y múltiples genes modificadores.
Mecanismo de desarrollo
Sección titulada «Mecanismo de desarrollo»La fisura embrionaria (fisura de la copa óptica) se forma ventralmente cuando la copa óptica se desarrolla a partir del neuroectodermo en la semana 4 de gestación. Se completa en la semana 5 y el cierre comienza en la semana 6. El cierre progresa desde cerca del ecuador hacia adelante (lado del iris) y hacia atrás (lado del nervio óptico), completándose en la semana 7. El cierre incompleto localizado en la parte posterior da lugar al coloboma del disco óptico.
Genes relacionados
Sección titulada «Genes relacionados»| Gen | Enfermedad asociada | Notas |
|---|---|---|
| PAX2 | Síndrome de coloboma renal (renal coloboma syndrome) | Participa en la determinación ventral del ojo y el cierre de la fisura embrionaria1) |
| CHD7 | Síndrome CHARGE | Cromosoma 8 (8q12.2), enfermedad designada como intratable |
| FZD5 | Coloboma sindrómico + microcórnea | Receptor de la vía de señalización Wnt2) |
Complicaciones sistémicas
Sección titulada «Complicaciones sistémicas»El coloboma del disco óptico puede asociarse con los siguientes síndromes sistémicos.
- Síndrome CHARGE: Síndrome de malformaciones múltiples que toma las iniciales de coloboma (C), defectos cardíacos (H), atresia de coanas (A), retraso del crecimiento (R), hipoplasia genital (G) y anomalías del oído (E). El gen causante es CHD7 y está designado como enfermedad intratable.
- Síndrome de Aicardi: Se caracteriza por agenesia del cuerpo calloso, epilepsia y retraso del desarrollo psicomotor. Es más frecuente en niñas. Los colobomas suelen aparecer como múltiples espacios en blanco (lagunas) en la coroides y la retina.
- Síndrome de coloboma renal: Causado por mutaciones en el gen PAX2. Se asocia con anomalías de la formación renal y del tracto urinario. Es necesario un seguimiento a largo plazo de la función renal. Se ha identificado una mutación de cambio de marco c.76delG en PAX2 en una familia con glomeruloesclerosis focal y segmentaria (GEFS), y el espectro fenotípico es más amplio de lo que se pensaba1).
El coloboma del disco óptico puede asociarse con síndromes sistémicos como el síndrome CHARGE (malformaciones múltiples por mutación de CHD7, enfermedad intratable designada), el síndrome de Aicardi (agenesia del cuerpo calloso, epilepsia, predominio femenino) y el síndrome de coloboma renal (mutación de PAX2, anomalías renales y del tracto urinario). En casos bilaterales o cuando hay hallazgos sistémicos, se recomienda la consulta pediátrica y el asesoramiento genético.
4. Diagnóstico y métodos de exploración
Sección titulada «4. Diagnóstico y métodos de exploración»Métodos diagnósticos
Sección titulada «Métodos diagnósticos»El diagnóstico es posible solo con los hallazgos oftalmoscópicos. Los puntos clave del diagnóstico son una depresión blanca bien definida predominantemente en la parte inferior del disco óptico y un curso vascular anómalo característico (múltiples vasos que se originan en el borde o dentro de la depresión). Para el diagnóstico definitivo se utilizan ecografía, resonancia magnética, tomografía computarizada y tomografía de coherencia óptica (OCT).
Aunque se dice que los casos esporádicos son frecuentes, también puede haber antecedentes familiares, por lo que es necesario indagar cuidadosamente sobre los antecedentes familiares.
Se requiere resonancia magnética o tomografía computarizada de la cabeza para buscar malformaciones intracraneales (como agenesia del cuerpo calloso). Se debe realizar una consulta pediátrica para confirmar la ausencia de complicaciones sistémicas como el síndrome CHARGE o el síndrome de Aicardi.
Exploraciones
Sección titulada «Exploraciones»| Exploración | Propósito |
|---|---|
| Examen de fondo de ojo (con dilatación) | Evaluación de la morfología del disco óptico, el curso vascular y el desprendimiento de retina |
| OCT (Tomografía de Coherencia Óptica) | Evaluación detallada de la estructura del disco óptico y la mácula |
| Ecografía (modo B) | Detección de desprendimiento de retina cuando el fondo de ojo es de mala visualización |
| Campimetría | Evaluación de patrones de defecto del campo visual (p. ej., defecto del campo superior) |
| RMN cerebral | Búsqueda de complicaciones del SNC como agenesia del cuerpo calloso y encefalocele |
| Ecografía renal | Cribado del síndrome de coloboma renal |
| Prueba genética | Búsqueda de mutaciones en PAX2, CHD7, etc. (en casos bilaterales o sindrómicos) |
Diagnóstico diferencial
Sección titulada «Diagnóstico diferencial»| Diagnóstico diferencial | Puntos clave de diferenciación |
|---|---|
| Síndrome de morning glory | Proliferación glial en el centro de la papila, vasos radiales. El coloboma presenta excavación inferior predominante sin proliferación glial. |
| Estafiloma peripapilar | Abombamiento posterior de la esclera que rodea la papila. El coloboma es un defecto de la propia papila. |
| PFV/PHPV del disco óptico (persistencia del vítreo primario hiperplásico) | Asociado con hebras vítreas y pliegues retinianos. Los hallazgos de fondo de ojo difieren del coloboma. |
| Megalopapila | Papila grande pero morfología casi normal. Sin excavación ni anomalías en el curso vascular.5) |
| Hipoplasia del nervio óptico | Papila pequeña (relación DM/DD ≥3.2). El coloboma presenta papila agrandada y excavada. |
| Atrofia óptica glaucomatosa | Agrandamiento progresivo de la excavación y elevación de la presión intraocular. El coloboma es no progresivo con PIO normal. |
5. Tratamiento estándar
Sección titulada «5. Tratamiento estándar»El coloboma del disco óptico en sí mismo es una anomalía estructural congénita sin tratamiento curativo. El tratamiento sintomático es el pilar, según la presencia y el tipo de complicaciones.
Observación
Sección titulada «Observación»Dado que es una anomalía congénita no progresiva, se continúa con la observación periódica del fondo de ojo si no hay complicaciones como desprendimiento seroso de retina. En la infancia, se recomienda la oftalmoscopia con dilatación cada 6 meses a 1 año.
Manejo del Desprendimiento Seroso de Retina
Sección titulada «Manejo del Desprendimiento Seroso de Retina»No existe un tratamiento establecido para el desprendimiento seroso de retina, y se ha informado resolución espontánea. Puede considerarse la observación durante varios meses. Si no hay mejoría tras la observación, se considera la intervención quirúrgica.
Se hipotetiza que el líquido vítreo ingresa al espacio subretiniano debido a anomalías estructurales en el área excavada, y también se ha sugerido la posibilidad de entrada de líquido cefalorraquídeo a través de la comunicación entre la excavación y el espacio subaracnoideo.
Manejo del Desprendimiento Regmatógeno de Retina
Sección titulada «Manejo del Desprendimiento Regmatógeno de Retina»Para el desprendimiento regmatógeno de retina, se realiza vitrectomía y fotocoagulación alrededor de la excavación. El pronóstico visual postoperatorio no es necesariamente bueno.
Se ha realizado la reinserción retiniana con pegamento de fibrina para el desprendimiento de retina relacionado con coloboma, y se ha informado una técnica de aplicación de pegamento de fibrina alrededor de los desgarros retinianos en el margen del coloboma para fortalecer la adhesión 3). Algunos casos han logrado una mejoría a una agudeza visual final de 20/50.
Tratamiento de la Ambliopía
Sección titulada «Tratamiento de la Ambliopía»Para casos con mala visión, especialmente unilateral, se realiza corrección refractiva y terapia de oclusión (parche del ojo sano). La intervención temprana en la infancia es importante. Sin embargo, el efecto del tratamiento de la ambliopía es limitado en casos de pérdida visual debida a anomalías estructurales del propio nervio óptico.
Manejo sistémico
Sección titulada «Manejo sistémico»- Casos con síndrome CHARGE: es necesario un manejo multidisciplinario que incluya cirugía cardíaca, otorrinolaringología, endocrinología, etc.
- Síndrome de coloboma renal: se realiza un seguimiento a largo plazo de la función renal.
- Consejo genético: recomendado en casos bilaterales o sindrómicos.
El desprendimiento seroso de retina puede resolverse espontáneamente, por lo que primero se realiza una observación durante varios meses. Para el desprendimiento regmatógeno, se realiza vitrectomía y fotocoagulación alrededor de la depresión. En los últimos años, también se han reportado casos de refuerzo de la adhesión con pegamento de fibrina. Sin embargo, el pronóstico visual postoperatorio no es necesariamente bueno.
6. Fisiopatología y patogenia detallada
Sección titulada «6. Fisiopatología y patogenia detallada»Proceso de cierre de la fisura embrionaria
Sección titulada «Proceso de cierre de la fisura embrionaria»La copa óptica se forma a partir del neuroectodermo en la cuarta semana de gestación. En la cara ventral de la copa óptica se forma una fisura embrionaria (fisura óptica), a través de la cual pasa la arteria hialoidea. Esta fisura se completa en la quinta semana y el cierre comienza en la sexta semana. El cierre comienza cerca del ecuador y progresa hacia adelante (hacia el iris) y hacia atrás (hacia el nervio óptico), completándose en la séptima semana. El fallo localizado del cierre posterior da lugar al coloboma del nervio óptico.
En el proceso de cierre participa la transición epitelio-mesenquimatosa (EMT). Las células epiteliales de la retina neural en los bordes de la fisura embrionaria degradan la membrana basal, adquieren características mesenquimatosas y se fusionan. La alteración de este proceso conduce al coloboma6).
Mecanismos moleculares
Sección titulada «Mecanismos moleculares»El gen PAX2 está implicado en la determinación ventral del ojo y se cree que participa en el cierre de la fisura embrionaria. Las mutaciones de PAX2 causan el síndrome de coloboma renal. Se ha identificado una mutación de cambio de marco c.76delG de PAX2 en una familia con FSGS, y el fenotipo del síndrome de coloboma renal es más amplio de lo que se pensaba1).
El gen FZD5 codifica un receptor de la vía de señalización Wnt. Las mutaciones con pérdida de función alteran la activación dependiente de ligando de la señalización Wnt, lo que provoca un cierre defectuoso de la fisura embrionaria y microcórnea. Sigue un patrón de herencia recesivo2).
El gen CHD7 codifica un factor de remodelación de la cromatina y participa en la diferenciación y migración de las células de la cresta neural. Las mutaciones causan el síndrome CHARGE.
Mecanismo del desprendimiento seroso de retina
Sección titulada «Mecanismo del desprendimiento seroso de retina»Se hipotetiza que el humor vítreo fluye hacia el espacio subretiniano debido a anomalías estructurales en el área del coloboma. También se ha sugerido la posibilidad de entrada de líquido cefalorraquídeo a través de la comunicación entre el coloboma y el espacio subaracnoideo, lo que se considera una de las razones por las que el tratamiento del desprendimiento seroso de retina es difícil.
Pronóstico y Evolución
Sección titulada «Pronóstico y Evolución»El coloboma del disco óptico es una anomalía congénita fija y no progresiva. La presencia de desprendimiento de retina es el principal factor que empeora el pronóstico visual. Incluso después de la vitrectomía para el desprendimiento regmatógeno de retina, el pronóstico visual suele ser malo.
7. Investigación más reciente y perspectivas futuras
Sección titulada «7. Investigación más reciente y perspectivas futuras»Ampliación del espectro fenotípico de las mutaciones de PAX2
Sección titulada «Ampliación del espectro fenotípico de las mutaciones de PAX2»Hu et al. (2024) informaron que se identificó una mutación de cambio de marco c.76delG en PAX2 en una familia con glomeruloesclerosis focal y segmentaria (GEFS) 1). El fenotipo del síndrome de coloboma renal es más amplio de lo que se pensaba anteriormente, y se ha reafirmado la importancia del cribado de la función renal en casos de coloboma del disco óptico. Se sugiere ampliar las indicaciones para la evaluación de la función renal en pacientes con coloboma.
FZD5 y anomalías en la señalización de Wnt
Sección titulada «FZD5 y anomalías en la señalización de Wnt»Cortes-Gonzalez et al. (2024) informaron que una mutación homocigótica de cambio de sentido en FZD5 (p.M160V) causa coloboma ocular sindrómico y microcórnea 2). El análisis funcional confirmó que la activación dependiente de ligando de la señalización de Wnt se ve afectada en un patrón de herencia recesivo. La tasa de diagnóstico genético del coloboma es inferior al 30%, y se espera que la identificación de nuevos genes causantes contribuya a mejorar el diagnóstico.
Nuevas técnicas quirúrgicas para el desprendimiento de retina
Sección titulada «Nuevas técnicas quirúrgicas para el desprendimiento de retina»Jain et al. (2024) informaron la realización de una cirugía de reaplicación retiniana con pegamento de fibrina para el desprendimiento de retina asociado a coloboma, logrando una mejoría hasta una agudeza visual final de 20/50 3). La técnica de aplicar pegamento de fibrina alrededor de los desgarros retinianos en el margen del coloboma para mejorar la adhesión está atrayendo la atención como una opción complementaria a la vitrectomía convencional más fotocoagulación. La acumulación de casos y la verificación de los resultados a largo plazo son desafíos futuros.
8. Referencias
Sección titulada «8. Referencias»-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.