
Coloboma do disco óptico
Pontos-chave em resumo
Seção intitulada “Pontos-chave em resumo”1. O que é coloboma do disco óptico?
Seção intitulada “1. O que é coloboma do disco óptico?”Coloboma do disco óptico é uma anomalia congênita caracterizada por alargamento anormal do disco óptico e uma depressão branca bem delimitada. Ocorre devido ao fechamento incompleto da fissura óptica (fissura embrionária), que se fecha na 7ª semana de gestação. Os vasos retinianos não se originam de um único ponto, mas de vários locais na borda ou dentro da depressão.
Quando o fechamento incompleto da fissura óptica se limita à parte posterior (lado do nervo óptico), ocorre o coloboma do nervo óptico. Se houver um fechamento incompleto mais extenso de anterior para posterior, forma-se um espectro de coloboma de íris–coroide. Dentro do espectro geral do coloboma, o coloboma do disco óptico corresponde à extremidade posterior do fechamento incompleto da fissura óptica e faz parte de um espectro contínuo desde o coloboma de íris (extremidade anterior), mas também existe uma forma localizada apenas no disco óptico.
O código CID-10 é H47.319 (nervo óptico).
É importante diferenciar da síndrome da glória-da-manhã. Na síndrome da glória-da-manhã, há tecido glial proliferativo no centro do disco óptico e os vasos se distribuem radialmente. No coloboma do disco óptico, não há proliferação glial, a depressão é mais proeminente inferiormente e os vasos se originam de vários locais na borda ou dentro da depressão, o que permite a diferenciação.
O coloboma do disco óptico apresenta uma depressão branca bem delimitada predominante inferiormente, e os vasos se originam de vários locais na borda ou dentro da depressão. Não há proliferação glial. Na síndrome da glória-da-manhã, há tecido glial proliferativo no centro do disco óptico e os vasos se distribuem radialmente a partir da periferia do disco. Ambas são anomalias congênitas do disco óptico, mas podem ser diferenciadas pelo exame de fundo de olho.
2. Principais sintomas e achados clínicos
Seção intitulada “2. Principais sintomas e achados clínicos”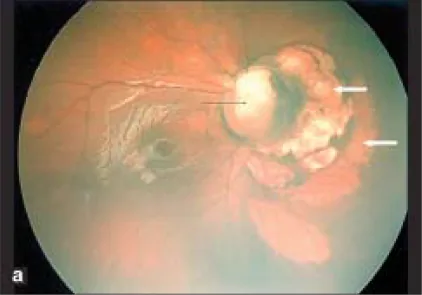
Sintomas subjetivos
Seção intitulada “Sintomas subjetivos”A acuidade visual é determinada pelo envolvimento do feixe papilomacular no coloboma e sua extensão. A acuidade varia de melhor que 1,0 a casos ruins, mas frequentemente a acuidade está reduzida mesmo sem dano macular devido à anomalia do nervo óptico.
- Baixa acuidade visual: Depende do grau de envolvimento do feixe papilomacular. Em casos de visão ruim, pode ocorrer estrabismo por ambliopia.
- Defeito de campo visual: Frequentemente ocorre defeito de campo visual superior correspondente ao coloboma na porção inferior do disco.
- Estrabismo: Em casos de visão ruim, pode ocorrer estrabismo por ambliopia secundário.
Achados de fundo de olho
Seção intitulada “Achados de fundo de olho”No fundo de olho, ocorre um defeito do disco óptico e da coriorretina na porção inferior do globo ocular. Acompanha-se de anomalia no trajeto dos vasos, e como a artéria central da retina se ramifica atrás do disco, muitas artérias retinianas parecem emergir do disco. A borda superior do disco frequentemente permanece, e mesmo quando todo o disco é escavado, tipicamente a porção inferior é mais profunda que a superior.
A região do disco é escavada, o disco está ausente ou parcialmente ausente, e a coroide circundante, o epitélio pigmentar da retina e a esclera são finos. Abaixo da escavação da região do disco, há atrofia coriorretiniana e aspecto estriado devido ao fechamento incompleto da fissura embrionária.
Classificação
Seção intitulada “Classificação”O coloboma do disco óptico é classificado de acordo com a extensão do envolvimento da seguinte forma.
- Tipo isolado de coloboma do disco óptico: Causado pelo fechamento incompleto localizado da porção posterior da fissura do cálice óptico.
- Tipo com coloboma coriorretiniano: Indica um fechamento incompleto mais extenso da fissura do cálice óptico.
- Tipo combinado de coloboma de íris e corpo ciliar: Tipo extenso contínuo da extremidade anterior à posterior.
- Coloboma de Fuchs: Tipo leve, mostrando lesão atrófica semelhante a um cone abaixo da papila. A visão é frequentemente preservada.
Complicações
Seção intitulada “Complicações”Complicações oculares
Seção intitulada “Complicações oculares”- Frequentemente associado a coloboma de íris e coloboma de coroide.
- Quando associado a coloboma de coroide e área de depressão ampla, pode apresentar microftalmia.
- Descolamento seroso de retina: Pode ocorrer mesmo com coloboma isolado do disco óptico.
- Descolamento regmatogênico de retina: Pode ocorrer secundariamente em casos de coloboma complexo de retina e coroide.
- Hipotonia por filtração transescleral: Foram relatados casos de vazamento de humor aquoso através do defeito escleral 7).
3. Causas, Epidemiologia, Fatores de Risco
Seção intitulada “3. Causas, Epidemiologia, Fatores de Risco”Epidemiologia
Seção intitulada “Epidemiologia”A prevalência é relatada como 3–8/100.000. Acredita-se que a unilateralidade e bilateralidade sejam equivalentes, a maioria dos casos é esporádica, mas frequentemente há histórico familiar. Vários padrões de herança foram relatados, como autossômico dominante, autossômico recessivo e ligado ao X 4).
A taxa de diagnóstico genético para todo o coloboma é inferior a 30% 5). Muitos casos esporádicos não têm mutações genéticas identificadas, e supõe-se o envolvimento de fatores ambientais e múltiplos genes modificadores.
Mecanismo de formação
Seção intitulada “Mecanismo de formação”A fissura embrionária (fissura do cálice óptico) ocorre na 4ª semana de gestação quando o cálice óptico se forma a partir do neuroectoderma no lado ventral. Completa-se na 5ª semana, e o fechamento começa na 6ª semana. O fechamento progride da região equatorial em direção anterior (íris) e posterior (nervo óptico), completando-se na 7ª semana. Se o fechamento posterior for incompleto e localizado, ocorre coloboma do nervo óptico.
Genes relacionados
Seção intitulada “Genes relacionados”| Gene | Doença associada | Observações |
|---|---|---|
| PAX2 | Síndrome do coloboma renal | Envolvido na determinação ventral do olho e fechamento da fissura embrionária1) |
| CHD7 | Síndrome CHARGE | Cromossomo 8 (8q12.2), doença rara designada |
| FZD5 | Coloboma sintomático + microcórnea | Receptor da via de sinalização Wnt2) |
Complicações sistêmicas
Seção intitulada “Complicações sistêmicas”O coloboma do disco óptico pode estar associado às seguintes síndromes sistêmicas.
- Síndrome CHARGE: Síndrome de malformações multiorgânicas cujo nome é formado pelas iniciais: Coloboma (C), Cardiopatia (H), Atresia de coanas (A), Retardo de crescimento (R), Hipoplasia genital (G), Anomalia de orelha (E). O gene causador é CHD7, e é reconhecida como doença rara designada.
- Síndrome de Aicardi: Caracterizada por agenesia do corpo caloso, epilepsia e atraso do desenvolvimento mental. Mais comum em meninas. O coloboma frequentemente se apresenta como múltiplas lacunas na coriorretina.
- Síndrome do coloboma renal: Causada por mutação no gene PAX2. Associada a malformações do trato urinário e renal. É necessário acompanhamento a longo prazo da função renal. A mutação frameshift c.76delG no PAX2 foi identificada em famílias com glomeruloesclerose segmentar focal (GESF), indicando que o espectro fenotípico é mais amplo do que se pensava anteriormente 1).
O coloboma do disco óptico pode estar associado a síndromes sistêmicas como Síndrome CHARGE (mutação CHD7, malformações multiorgânicas, doença rara designada), Síndrome de Aicardi (agenesia do corpo caloso, epilepsia, predominância feminina) e Síndrome do coloboma renal (mutação PAX2, malformações renais e do trato urinário). Em casos bilaterais ou com achados sistêmicos, recomenda-se consulta pediátrica e aconselhamento genético.
4. Diagnóstico e Métodos de Exame
Seção intitulada “4. Diagnóstico e Métodos de Exame”Métodos de Diagnóstico
Seção intitulada “Métodos de Diagnóstico”O diagnóstico é possível apenas pela oftalmoscopia. Uma escavação branca de bordas nítidas na porção inferior do disco e anormalidades características no trajeto dos vasos (origem de múltiplos vasos na borda ou dentro da escavação) são os pontos-chave do diagnóstico. Para confirmação, utiliza-se ultrassonografia, ressonância magnética, tomografia computadorizada e tomografia de coerência óptica (OCT).
Embora a maioria dos casos seja esporádica, pode haver histórico familiar, sendo necessária uma anamnese cuidadosa do histórico familiar.
Para detectar malformações intracranianas associadas (como agenesia do corpo caloso), é necessária ressonância magnética ou tomografia computadorizada de crânio. A consulta pediátrica é realizada para verificar a presença de complicações sistêmicas como Síndrome CHARGE ou Síndrome de Aicardi.
| Exame | Objetivo |
|---|---|
| Exame de fundo de olho (com dilatação pupilar) | Avaliação da morfologia do disco óptico, trajeto vascular e descolamento de retina |
| OCT (Tomografia de Coerência Óptica) | Avaliação detalhada da estrutura do disco óptico e mácula |
| Ultrassonografia (modo B) | Pesquisa de descolamento de retina em casos com má visualização do fundo |
| Campimetria | Avaliação do padrão de defeito de campo (ex.: defeito de campo superior) |
| Ressonância magnética de crânio | Pesquisa de complicações do SNC como agenesia do corpo caloso e encefalocele |
| Ultrassonografia renal | Triagem para síndrome de coloboma renal |
| Teste genético | Pesquisa de mutações em PAX2, CHD7, etc. (em casos bilaterais e sindrômicos) |
Diagnóstico diferencial
Seção intitulada “Diagnóstico diferencial”| Doenças de Diferenciação | Pontos de Diferenciação |
|---|---|
| Síndrome da glória da manhã (morning glory syndrome) | Tecido glial no centro da papila, vasos em disposição radial. No coloboma, depressão predominante inferior e sem proliferação glial |
| Estafiloma peripapilar (peripapillary staphyloma) | Protrusão posterior da esclera ao redor da papila. Coloboma é um defeito na própria papila |
| PFV/PHPV papilar (persistência da artéria hialoide primitiva) | Acompanhado por cordão vítreo e prega retiniana. Difere do coloboma na aparência do fundo de olho |
| Megalopapila (megalopapilla) | Diâmetro da papila aumentado, mas forma próxima do normal. Sem depressão ou anormalidade no trajeto vascular5) |
| Hipoplasia do nervo óptico | Papila pequena (razão DM/DD ≥ 3,2). Coloboma causa papila aumentada e deprimida |
| Atrofia óptica glaucomatosa | Escavação progressiva e aumento da pressão intraocular. Coloboma não progressivo com pressão intraocular normal |
5. Métodos de tratamento padrão
Seção intitulada “5. Métodos de tratamento padrão”O coloboma do disco óptico é uma anomalia estrutural congênita e não há tratamento curativo. O tratamento é sintomático, dependendo da presença e do tipo de complicações.
Observação
Seção intitulada “Observação”Por ser uma anomalia congênita não progressiva, se não houver complicações como descolamento seroso da retina, a observação fundoscópica regular é continuada. Na infância, recomenda-se exame oftalmoscópico com dilatação a cada 6 meses a 1 ano.
Abordagem do descolamento seroso da retina
Seção intitulada “Abordagem do descolamento seroso da retina”Não há consenso sobre o tratamento do descolamento seroso da retina, e casos de resolução espontânea foram relatados. A observação por alguns meses pode ser realizada. Se não houver melhora após a observação, considera-se a intervenção cirúrgica.
Supõe-se que a anormalidade estrutural da escavação permita a entrada do humor vítreo no espaço sub-retiniano, e também é sugerida a possibilidade de entrada de líquido cefalorraquidiano através da comunicação entre a escavação e o espaço subaracnóideo.
Abordagem do descolamento regmatogênico da retina
Seção intitulada “Abordagem do descolamento regmatogênico da retina”Para o descolamento regmatogênico da retina, realiza-se vitrectomia e fotocoagulação ao redor da escavação. O prognóstico visual pós-operatório não é necessariamente bom.
O uso de cola de fibrina para reposicionamento da retina foi relatado em descolamentos relacionados a coloboma, onde a cola de fibrina é aplicada ao redor da ruptura retiniana na borda do coloboma para reforçar a adesão 3). Alguns casos mostraram melhora da acuidade visual final para 20/50.
Tratamento da ambliopia
Seção intitulada “Tratamento da ambliopia”Em casos de baixa visão, especialmente unilateral, realiza-se correção refrativa e oclusão (tamponamento do olho bom). A intervenção precoce na infância é importante. No entanto, na baixa visão devido à anomalia estrutural do próprio nervo óptico, o efeito do tratamento da ambliopia é limitado.
Manejo Sistêmico
Seção intitulada “Manejo Sistêmico”- Casos com síndrome CHARGE: requerem manejo multidisciplinar com cirurgia cardíaca, otorrinolaringologia, endocrinologia, etc.
- Síndrome renal-coloboma: realizar acompanhamento de longo prazo da função renal.
- Aconselhamento genético: recomendado em casos bilaterais ou sindrômicos.
O descolamento seroso de retina pode regredir espontaneamente, portanto, primeiro realiza-se observação por alguns meses. Para descolamento regmatogênico, realiza-se vitrectomia e fotocoagulação ao redor da área de depressão. Recentemente, há relatos de reforço da adesão com uso de cola de fibrina. No entanto, o prognóstico visual pós-operatório nem sempre é bom.
6. Fisiopatologia e Mecanismo Detalhado
Seção intitulada “6. Fisiopatologia e Mecanismo Detalhado”Processo de Fechamento da Fenda Embrionária
Seção intitulada “Processo de Fechamento da Fenda Embrionária”A taça óptica forma-se a partir do neuroectoderma na 4ª semana gestacional. Uma fenda embrionária (fenda da taça óptica) surge no lado ventral da taça óptica, por onde passa a artéria hialoide. Essa fenda completa-se na 5ª semana, e o fechamento inicia-se na 6ª semana. O fechamento começa próximo ao equador e progride anteriormente (em direção à íris) e posteriormente (em direção ao nervo óptico), completando-se na 7ª semana. O fechamento posterior incompleto localizado resulta em coloboma do nervo óptico.
O processo de fechamento envolve a transição epitélio-mesenquimal (EMT). As células epiteliais neurosensoriais da retina na borda da fenda embrionária degradam a membrana basal, adquirem fenótipo mesenquimal e fundem-se. A falha nesse processo causa coloboma6).
Mecanismo Molecular
Seção intitulada “Mecanismo Molecular”O gene PAX2 está envolvido na determinação ventral do olho e acredita-se que participe do fechamento da fenda embrionária. Mutações no PAX2 causam síndrome renal-coloboma. A mutação frameshift c.76delG no PAX2 foi identificada em uma família com FSGS, e o fenótipo da síndrome renal-coloboma é mais amplo do que se pensava anteriormente1).
O gene FZD5 codifica um receptor da via de sinalização Wnt. Mutações de perda de função prejudicam a ativação dependente de ligante da sinalização Wnt, resultando em fechamento incompleto da fenda embrionária e microcórnea. Segue padrão de herança recessiva2).
O gene CHD7 codifica um fator de remodelação da cromatina e está envolvido na diferenciação e migração das células da crista neural. Mutações causam síndrome CHARGE.
Mecanismo do Descolamento Seroso de Retina
Seção intitulada “Mecanismo do Descolamento Seroso de Retina”Supõe-se que anormalidades estruturais na área de escavação levem ao fluxo de humor vítreo para o espaço sub-retiniano. Também foi sugerida a possibilidade de influxo de líquido cefalorraquidiano através da comunicação entre a escavação e o espaço subaracnóideo, sendo considerada uma das razões para a dificuldade de tratamento do descolamento seroso da retina.
Prognóstico e Evolução
Seção intitulada “Prognóstico e Evolução”O coloboma do disco óptico é uma anomalia congênita fixa e não progressiva. A complicação do descolamento de retina é o principal fator que piora o prognóstico visual. Mesmo após a vitrectomia para descolamento regmatogênico da retina, o prognóstico visual muitas vezes não é favorável.
7. Pesquisas Recentes e Perspectivas Futuras
Seção intitulada “7. Pesquisas Recentes e Perspectivas Futuras”Expansão do Espectro Fenotípico de Mutações PAX2
Seção intitulada “Expansão do Espectro Fenotípico de Mutações PAX2”Hu et al. (2024) relataram a identificação de uma mutação de deslocamento de quadro c.76delG no PAX2 em uma família com glomeruloesclerose segmentar focal (GESF) 1). O espectro fenotípico da síndrome do coloboma renal é mais amplo do que se pensava anteriormente, e a importância da triagem da função renal em casos de coloboma do disco óptico foi novamente enfatizada. Sugere-se ampliar a indicação de avaliação da função renal em pacientes com coloboma.
FZD5 e Anormalidade da Sinalização Wnt
Seção intitulada “FZD5 e Anormalidade da Sinalização Wnt”Cortes-Gonzalez et al. (2024) relataram que uma mutação missense homozigótica no FZD5 (p.M160V) causa coloboma ocular sintomático e microcórnea 2). A análise funcional confirmou que o padrão de herança recessiva prejudica a ativação da sinalização Wnt dependente de ligante. A taxa de diagnóstico genético do coloboma é inferior a 30%, e espera-se que a identificação de novos genes causadores contribua para a melhoria do diagnóstico.
Novas Técnicas Cirúrgicas para Descolamento de Retina
Seção intitulada “Novas Técnicas Cirúrgicas para Descolamento de Retina”Jain et al. (2024) relataram a realização de reposicionamento retiniano com cola de fibrina para descolamento de retina associado a coloboma, com melhora da acuidade visual final para 20/50 3). A técnica de aplicação de cola de fibrina ao redor da ruptura retiniana na borda do coloboma para fortalecer a adesão é uma opção adjuvante promissora para a vitrectomia convencional com fotocoagulação. O acúmulo de casos e a verificação dos resultados a longo prazo são desafios futuros.
8. Referências
Seção intitulada “8. Referências”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.