
Optik Disk Kolobomu
Bir bakışta önemli noktalar
Section titled “Bir bakışta önemli noktalar”1. Optik Disk Kolobomu Nedir?
Section titled “1. Optik Disk Kolobomu Nedir?”Optik disk kolobomu, optik diskte anormal genişleme ve sınırları net beyaz çöküntü ile karakterize konjenital bir anomalidir. Embriyonik 7. haftada kapanması gereken optik fissürün (embriyonik yarık) kapanma defekti (kolobom) sonucu oluşur. Retina damarları tek bir noktadan çıkmaz, çöküntü kenarı veya içindeki çeşitli noktalardan çıkarlar.
Optik fissürün kapanma defekti arka kısımda (optik disk tarafında) sınırlı olduğunda optik disk kolobomu oluşur. Önden arkaya daha yaygın bir kapanma defekti varsa, iris-koroid kolobomu spektrumu oluşur. Tüm kolobom spektrumunda optik disk kolobomu, optik fissür kapanma defektinin arka ucuna karşılık gelir ve iris kolobomundan (ön uç) devam eden spektrumun bir parçasıdır, ancak optik disk ile sınırlı lokalize tip de mevcuttur.
ICD-10 kodu H47.319’dur (optik sinir).
Sabah zaferi sendromundan ayırt edilmesi önemlidir. Sabah zaferi sendromunda disk merkezinde glial proliferasyon dokusu görülür ve damarlar radyal olarak uzanır. Optik disk kolobomunda glial proliferasyon yoktur, çöküntü alt kadranda belirgindir ve damarlar çöküntü kenarı veya içindeki çeşitli noktalardan çıkar; bu özelliklerle ayırt edilebilir.
Optik disk kolobomu, alt kadranda belirgin, sınırları net beyaz çöküntü ile karakterizedir ve damarlar çöküntü kenarı veya içindeki çeşitli noktalardan çıkar. Glial proliferasyon dokusu görülmez. Sabah zaferi sendromunda disk merkezinde glial proliferasyon dokusu bulunur ve damarlar disk kenarından radyal olarak uzanır. Her ikisi de optik diskin konjenital anomalileridir, ancak fundus bulguları ile ayırt edilebilirler.
2. Ana belirtiler ve klinik bulgular
Section titled “2. Ana belirtiler ve klinik bulgular”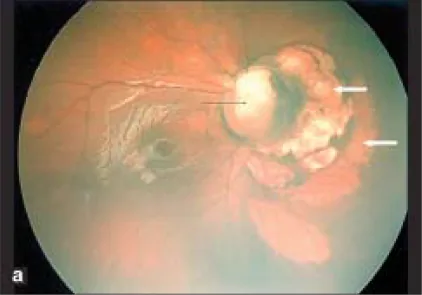
Subjektif belirtiler
Section titled “Subjektif belirtiler”Görme keskinliği, papillomaküler lif demetinin koloboma dahil olup olmadığına ve derecesine bağlıdır. 1.0’ın üzerinden kötü vakalara kadar değişen dereceler olmakla birlikte, optik sinir anomalisi nedeniyle makula sağlam olsa bile birçok vakada görme keskinliği azalmıştır.
- Görme azalması: Papillomaküler lif demetinin tutulum derecesine bağlıdır. Görme keskinliği kötü olan vakalarda kullanılmama şaşılığı (ambliyopiye bağlı şaşılık) görülebilir.
- Görme alanı defekti: Diskin alt kısmındaki koloboma karşılık gelen üst görme alanı defekti sıklıkla oluşur.
- Şaşılık: Görme keskinliği kötü olan vakalarda kullanılmama şaşılığı sekonder olarak gelişir.
Fundus bulguları
Section titled “Fundus bulguları”Fundusta, optik disk ve göz küresinin alt kısmında koroid ve retinada, alt kadran ağırlıklı bir defekt oluşur. Damar seyir anomalileri eşlik eder ve santral retinal arter disk arkasında dallandığı için, diskten çok sayıda retinal arter çıkıyormuş gibi görünür. Diskin üst kenarı sıklıkla korunur ve tüm disk çökük olsa bile tipik olarak üst kısma kıyasla alt kısım daha belirgindir.
Disk bölgesi çöküktür, disk yoktur veya kısmi defektlidir ve çevredeki koroid, retina pigment epiteli (RPE) ve sklera da incelmiştir. Disk bölgesindeki çöküntünün altında, fetal fissürün kapanma yetersizliğine bağlı koroid ve retina atrofisi ve mozaik görünümü mevcuttur.
Sınıflandırma
Section titled “Sınıflandırma”Optik disk kolobomu, eşlik eden tutulum alanına göre aşağıdaki gibi sınıflandırılır.
- İzole optik disk kolobomu tipi: Optik kese yarığının arka kısmının lokalize kapanma yetersizliğine bağlıdır.
- Koroid ve retina kolobomu ile birlikte olan tip: Daha geniş bir optik kese yarığı kapanma yetersizliğini gösterir.
- İris ve siliyer cisim kolobomu ile birlikte olan tip: Önden arkaya kadar sürekli olan yaygın tip.
- Fuchs kolobomu: Hafif tiptir ve optik disk altında konusa benzer atrofik lezyon gösterir. Görme genellikle nispeten korunur.
Komplikasyonlar
Section titled “Komplikasyonlar”Göz komplikasyonları
Section titled “Göz komplikasyonları”- Sıklıkla iris kolobomu ve koroid kolobomu ile birliktedir.
- Koroid kolobomu eşlik ediyorsa ve çöküntü alanı genişse mikroftalmi görülebilir.
- Seröz retina dekolmanı: İzole optik disk kolobomunda bile görülebilir.
- Regmatojen retina dekolmanı: Karmaşık koroidoretinal kolobom olgularında sekonder olarak gelişebilir.
- Transskleral filtrasyona bağlı hipotoni: Sklera defektinden aköz sızıntısı olan olgular bildirilmiştir7).
3. Nedenler, Epidemiyoloji, Risk Faktörleri
Section titled “3. Nedenler, Epidemiyoloji, Risk Faktörleri”Epidemiyoloji
Section titled “Epidemiyoloji”Prevalans 3-8/100.000 olarak bildirilmiştir. Tek taraflı ve çift taraflı olgular eşit sıklıktadır ve çoğu sporadiktir, ancak sıklıkla aile öyküsü vardır. Otozomal dominant, otozomal resesif ve X’e bağlı gibi çeşitli kalıtım şekilleri bildirilmiştir4).
Tüm kolobomların genetik tanı oranı %30’un altındadır5). Genetik mutasyon saptanamayan birçok sporadik olgu vardır ve çevresel faktörler veya birden fazla modifiye edici genin rol oynadığı düşünülmektedir.
Oluşum mekanizması
Section titled “Oluşum mekanizması”Embriyonik yarık (optik yarık), embriyonun 4. haftasında nöroektodermden optik kese oluşurken ventralde meydana gelir. 5. haftada tamamlanır ve 6. haftadan itibaren kapanma başlar. Kapanma, ekvator yakınından öne (iris tarafına) ve arkaya (optik sinir tarafına) doğru ilerler ve 7. haftada tamamlanır. Arka kapanmanın sınırlı bir alanda eksik olması optik disk kolobomuna yol açar.
İlgili Genler
Section titled “İlgili Genler”| Gen | İlgili Hastalık | Açıklama |
|---|---|---|
| PAX2 | Renal kolobom sendromu | Gözün ventral belirlenmesi ve fetal fissürün kapanmasında rol oynar1) |
| CHD7 | CHARGE sendromu | Kromozom 8 (8q12.2), belirlenmiş nadir hastalık |
| FZD5 | Semptomatik kolobom + mikrokornea | Wnt sinyal yolu reseptörü2) |
Sistemik Komplikasyonlar
Section titled “Sistemik Komplikasyonlar”Optik disk kolobomu aşağıdaki sistemik sendromlarla birlikte görülebilir.
- CHARGE sendromu: Kolobom (C), kalp anomalisi (H), koanal atrezi (A), büyüme geriliği (R), genital hipoplazi (G) ve dış kulak anomalisi (E) kelimelerinin baş harflerinden oluşan çoklu organ malformasyon sendromu. CHD7 geni sorumlu gendir ve nadir hastalık olarak tanımlanmıştır.
- Aicardi sendromu: Korpus kallozum agenezisi, epilepsi ve zihinsel gelişim geriliği ile birliktedir. Kızlarda daha sık görülür. Kolobom genellikle koroid ve retinada çok sayıda laküna olarak ortaya çıkar.
- Renal kolobom sendromu: PAX2 gen mutasyonuna bağlıdır. Renal üriner sistem anomalileri eşlik eder. Uzun süreli böbrek fonksiyon takibi gereklidir. PAX2 c.76delG çerçeve kayması mutasyonu, fokal segmental glomerüloskleroz (FSGS) olan bir ailede tanımlanmıştır ve fenotipik spektrum önceden düşünülenden daha geniştir 1).
CHARGE sendromu (CHD7 mutasyonuna bağlı çoklu organ malformasyonu, nadir hastalık), Aicardi sendromu (korpus kallozum agenezisi, epilepsi, kızlarda baskın) ve renal kolobom sendromu (PAX2 mutasyonu, renal üriner sistem anomalileri) gibi sistemik sendromlar eşlik edebilir. Bilateral olgularda veya sistemik bulgular varlığında pediatri konsültasyonu ve genetik danışmanlık önerilir.
4. Tanı ve test yöntemleri
Section titled “4. Tanı ve test yöntemleri”Tanı yöntemi
Section titled “Tanı yöntemi”Tanı sadece oftalmoskopik bulgularla konulabilir. Diskin alt kısmında belirgin sınırlı beyaz çöküntü ve karakteristik vasküler anormallik (çöküntü kenarından veya içinden çok sayıda damar çıkışı) tanının ana noktalarıdır. Kesin tanı için ultrason, MRI, BT ve optik koherens tomografi (OCT) kullanılır.
Sporadik vakalar daha sık olmakla birlikte aile öyküsü de olabilir, bu nedenle dikkatli bir aile öyküsü alınması gereklidir.
İntrakraniyal anomalilerin (korpus kallozum agenezisi gibi) araştırılması için kafa MRI/BT gereklidir. CHARGE sendromu, Aicardi sendromu gibi sistemik komplikasyonları ekarte etmek için pediatri konsültasyonu yapılır.
Testler
Section titled “Testler”| Test | Amaç |
|---|---|
| Fundus muayenesi (pupil genişletilerek) | Optik disk şekli, damar seyri ve retina dekolmanının değerlendirilmesi |
| OCT (Optik Koherens Tomografi) | Optik disk ve makula yapısının ayrıntılı değerlendirilmesi |
| Ultrasonografi (B-mod) | Fundusun iyi görülemediği durumlarda retina dekolmanı araştırılması |
| Görme alanı testi | Görme alanı defekti paterninin değerlendirilmesi (üst kadran defekti gibi) |
| Kranial MRG | Korpus kallozum agenezisi, ensefalosel gibi santral sinir sistemi komplikasyonlarının araştırılması |
| Böbrek ultrasonu | Renal kolobom sendromu taraması |
| Genetik test | PAX2, CHD7 vb. mutasyonlarının araştırılması (bilateral ve sendromik olgularda) |
Ayırıcı tanı
Section titled “Ayırıcı tanı”| Ayırıcı tanı hastalıkları | Ayırıcı noktalar |
|---|---|
| Sabah sefası sendromu (morning glory syndrome) | Optik disk merkezinde glial proliferasyon, damarlar radyal seyirli. Kolobomda alt kadranda çöküntü baskın, glial proliferasyon yok |
| Peripapiller stafilom (peripapillary staphyloma) | Optik diski çevreleyen skleranın arka kabarıklığı. Kolobom diskin kendisinin defektidir |
| Optik disk PFV/PHPV (primer vitreus arter kalıntısı) | Vitreus kordonu ve retina kıvrımı eşlik eder. Fundus bulguları kolobomdan farklıdır |
| Megalopapilla (megalopapilla) | Disk çapı büyük ancak şekil normale yakın. Çöküntü veya damar seyir anormalliği yok 5) |
| Optik sinir hipoplazisi | Disk küçük (DM/DD oranı ≥ 3.2). Kolobomda disk genişlemiş ve çöküntülüdür |
| Glokomatöz optik atrofi | Progresif çöküntü genişlemesi ve göz içi basıncı yüksekliği. Kolobom non-progresiftir ve göz içi basıncı normaldir |
5. Standart tedavi yöntemleri
Section titled “5. Standart tedavi yöntemleri”Optik disk kolobomu doğuştan gelen yapısal bir anomalidir ve kesin bir tedavisi yoktur. Tedavi, komplikasyonların varlığına ve türüne göre semptomatik olarak yapılır.
İlerleyici olmayan doğuştan bir anomali olduğu için, seröz retina dekolmanı gibi komplikasyonlar yoksa düzenli fundus muayenelerine devam edilir. Çocukluk döneminde altı ayda bir veya yılda bir dilatasyonlu oftalmoskopi önerilir.
Seröz retina dekolmanına yaklaşım
Section titled “Seröz retina dekolmanına yaklaşım”Seröz retina dekolmanının tedavisi konusunda fikir birliği yoktur ve kendiliğinden düzelen vakalar bildirilmiştir. Bazen birkaç ay izlem yapılır. İzlem sonrası düzelme olmazsa cerrahi müdahale düşünülür.
Çöküntü bölgesindeki yapısal anomali nedeniyle vitreus sıvısının subretinal alana kaçtığı düşünülmekte ve çöküntü ile subaraknoid boşluk arasındaki bağlantı yoluyla BOS kaçağı olasılığı da öne sürülmektedir.
Regmatojen retina dekolmanına yaklaşım
Section titled “Regmatojen retina dekolmanına yaklaşım”Regmatojen retina dekolmanında vitrektomi ve çöküntü çevresine fotokoagülasyon uygulanır. Cerrahi sonrası görme prognozu her zaman iyi değildir.
Fibrin yapıştırıcı kullanılarak retina redüksiyonu, kolobom ilişkili retina dekolmanında uygulanmış ve kolobom kenarındaki retina yırtığı çevresine fibrin yapıştırıcı sürülerek yapışmanın güçlendirildiği bir yöntem bildirilmiştir 3). Bazı vakalarda son görme keskinliği 20/50’ye düzelmiştir.
Ambliyopi tedavisi
Section titled “Ambliyopi tedavisi”Görme azlığı olan, özellikle tek taraflı vakalarda refraksiyon düzeltmesi ve kapama tedavisi (sağlam gözü kapatma) uygulanır. Çocukluk döneminde erken müdahale önemlidir. Ancak optik sinirin yapısal anomalisine bağlı görme azlığında ambliyopi tedavisinin etkinliği sınırlıdır.
Sistemik yönetim
Section titled “Sistemik yönetim”- CHARGE sendromu birlikteliği: Kalp cerrahisi, kulak burun boğaz, endokrinoloji vb. ile multidisipliner yönetim gereklidir.
- Renal kolobom sendromu: Böbrek fonksiyonlarının uzun süreli takibi yapılır.
- Genetik danışmanlık: Bilateral veya sendromik vakalarda önerilir.
Seröz retina dekolmanı bazen kendiliğinden gerileyebileceğinden, önce birkaç ay gözlem yapılır. Regmatojen dekolmanda vitrektomi ve çökertme çevresine fotokoagülasyon uygulanır. Son yıllarda fibrin yapıştırıcı ile yapışmayı güçlendirme raporları da vardır. Ancak ameliyat sonrası görme prognozu her zaman iyi değildir.
6. Patofizyoloji ve ayrıntılı oluşum mekanizması
Section titled “6. Patofizyoloji ve ayrıntılı oluşum mekanizması”Embriyonik yarığın kapanma süreci
Section titled “Embriyonik yarığın kapanma süreci”Optik kadeh, embriyonik 4. haftada nöroektodermden oluşur. Optik kadehin ventral tarafında embriyonik yarık (optik fissür) oluşur ve hyaloid arter buradan geçer. Bu yarık 5. haftada tamamlanır ve 6. haftadan itibaren kapanmaya başlar. Kapanma ekvator yakınından başlayarak öne (iris tarafı) ve arkaya (optik sinir tarafı) doğru ilerler ve 7. haftada tamamlanır. Arka kısmın sınırlı kapanma yetmezliği optik sinir kolobomuna yol açar.
Kapanma sürecinde epitelyal-mezenkimal geçiş (EMT) rol oynar. Embriyonik yarık kenarındaki nöral retina epitel hücreleri bazal membranı parçalayarak mezenkimal özellik kazanır ve birleşir. Bu sürecin bozulması koloboma neden olur6).
Moleküler mekanizma
Section titled “Moleküler mekanizma”PAX2 geni gözün ventral belirlenmesinde rol oynar ve embriyonik yarığın kapanmasında görev aldığı düşünülmektedir. PAX2 mutasyonu renal kolobom sendromuna neden olur. PAX2’nin c.76delG çerçeve kayması mutasyonu FSGS’li bir ailede tanımlanmıştır ve renal kolobom sendromunun fenotipi önceden düşünülenden daha geniştir1).
FZD5 geni Wnt sinyal yolunun reseptörünü kodlar. İşlev kaybı mutasyonları, Wnt sinyalinin ligand bağımlı aktivasyonunu bozarak embriyonik yarık kapanma yetmezliği ve mikrokorneaya yol açar. Otozomal resesif kalıtım gösterir2).
CHD7 geni kromatin yeniden şekillenme faktörünü kodlar ve nöral krest hücrelerinin farklılaşması ve göçünde rol oynar. Mutasyonu CHARGE sendromuna neden olur.
Seröz retina dekolmanının mekanizması
Section titled “Seröz retina dekolmanının mekanizması”Çöküntü bölgesindeki yapısal anormallik nedeniyle vitreus sıvısının subretinal boşluğa sızdığı varsayılmaktadır. Ayrıca, çöküntü ile subaraknoid boşluk arasındaki bağlantı yoluyla BOS sızıntısı olasılığı da öne sürülmüştür ve bu durum seröz retina dekolmanı tedavisini zorlaştıran faktörlerden biri olarak kabul edilir.
Prognoz ve Seyir
Section titled “Prognoz ve Seyir”Optik disk kolobomu, ilerleyici olmayan, sabit bir konjenital anomalidir. Retina dekolmanı komplikasyonu, görme prognozunu kötüleştiren ana faktördür. Regmatojen retina dekolmanı için vitrektomi sonrasında bile görme prognozu genellikle umut verici değildir.
7. Güncel Araştırmalar ve Gelecek Perspektifleri
Section titled “7. Güncel Araştırmalar ve Gelecek Perspektifleri”PAX2 Mutasyonunun Fenotipik Spektrumunun Genişlemesi
Section titled “PAX2 Mutasyonunun Fenotipik Spektrumunun Genişlemesi”Hu ve ark. (2024), PAX2 genindeki c.76delG çerçeve kayması mutasyonunun fokal segmental glomerüloskleroz (FSGS) olan bir ailede tanımlandığını bildirmiştir 1). Renal kolobom sendromunun fenotipi daha önce düşünülenden daha geniştir ve optik disk kolobomu vakalarında böbrek fonksiyon taramasının önemi yeniden vurgulanmıştır. Kolobomu olan hastalarda böbrek fonksiyon değerlendirmesinin kapsamının genişletilmesi önerilmektedir.
FZD5 ve Wnt Sinyal İletim Anomalileri
Section titled “FZD5 ve Wnt Sinyal İletim Anomalileri”Cortes-Gonzalez ve ark. (2024), FZD5 genindeki homozigot yanlış anlamlı mutasyonun (p.M160V) semptomatik oküler kolobom ve mikrokorneaya neden olduğunu bildirmiştir 2). Fonksiyonel analiz, resesif kalıtım paterniyle Wnt sinyalinin ligand bağımlı aktivasyonunun bozulduğunu doğrulamıştır. Kolobomun genetik tanı oranı %30’un altındadır ve yeni nedensel genlerin tanımlanmasının tanıyı iyileştirmeye katkıda bulunması beklenmektedir.
Retina Dekolmanı için Yeni Cerrahi Teknikler
Section titled “Retina Dekolmanı için Yeni Cerrahi Teknikler”Jain ve ark. (2024), kolobom ilişkili retina dekolmanında fibrin yapıştırıcı ile retina restorasyonu uygulayarak son görme keskinliğinde 20/50’ye iyileşme bildirmiştir 3). Kolobom kenarındaki retina yırtığı çevresine fibrin yapıştırıcı uygulanarak yapışmanın güçlendirilmesi, geleneksel vitrektomi ve fotokoagülasyona yardımcı bir seçenek olarak dikkat çekmektedir. Vaka sayısının birikmesi ve uzun dönem sonuçlarının değerlendirilmesi gelecekteki zorluklardır.
8. Kaynaklar
Section titled “8. Kaynaklar”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.