
Coloboma della papilla ottica
Punti chiave a colpo d’occhio
Sezione intitolata “Punti chiave a colpo d’occhio”1. Cos’è il coloboma del disco ottico?
Sezione intitolata “1. Cos’è il coloboma del disco ottico?”Il coloboma del disco ottico è un’anomalia congenita caratterizzata da un allargamento anomalo del disco ottico e da una depressione bianca ben delimitata. Deriva da una chiusura incompleta della fessura della coppa ottica (fessura embrionale), che normalmente si chiude alla 7a settimana di gestazione. I vasi retinici non originano da un unico punto, ma da vari punti sul bordo o all’interno della depressione.
Quando la chiusura incompleta della fessura della coppa ottica è limitata alla parte posteriore (lato del nervo ottico), si verifica un coloboma del nervo ottico. Se la chiusura incompleta è più estesa dall’avanti all’indietro, forma uno spettro dal coloboma dell’iride a quello della coroide. Nell’intero spettro dei colobomi, il coloboma del disco ottico corrisponde all’estremità posteriore della chiusura incompleta della fessura della coppa ottica e fa parte dello spettro continuo con il coloboma dell’iride (estremità anteriore), ma esiste anche una forma localizzata isolata del disco ottico.
Il codice ICD-10 è H47.319 (nervo ottico).
La differenziazione dalla sindrome del morning glory è importante. Nella sindrome del morning glory si osserva un tessuto di proliferazione gliale al centro del disco, con vasi che decorrono radialmente. Nel coloboma del disco ottico non c’è proliferazione gliale, la depressione è predominante in basso e i vasi originano da vari punti sul bordo o all’interno della depressione, consentendo la differenziazione.
Il coloboma del disco ottico presenta una depressione bianca ben delimitata predominante in basso, e i vasi originano da vari punti sul bordo o all’interno della depressione. Non c’è tessuto di proliferazione gliale. Nella sindrome del morning glory, c’è un tessuto di proliferazione gliale al centro del disco e i vasi si irradiano radialmente dalla periferia del disco. Entrambi sono anomalie congenite del disco ottico, ma possono essere differenziati all’esame del fondo oculare.
2. Principali sintomi e segni clinici
Sezione intitolata “2. Principali sintomi e segni clinici”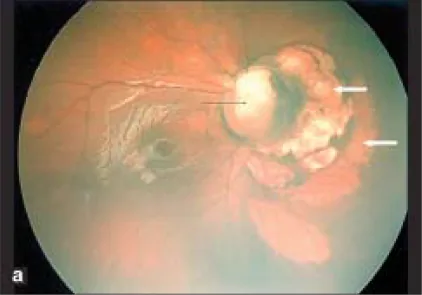
Sintomi soggettivi
Sezione intitolata “Sintomi soggettivi”L’acuità visiva dipende dal coinvolgimento del fascio papillomaculare nel coloboma e dalla sua entità. Varia da oltre 1,0 a casi di scarsa acuità, ma a causa dell’anomalia del nervo ottico, l’acuità visiva è spesso ridotta anche in assenza di danno maculare.
- Riduzione dell’acuità visiva: dipende dal grado di coinvolgimento del fascio papillomaculare. Nei casi di scarsa acuità, può verificarsi strabismo da non uso.
- Difetto del campo visivo: spesso si verifica un difetto del campo visivo superiore, corrispondente al coloboma inferiore della papilla.
- Strabismo: nei casi di scarsa acuità visiva, può svilupparsi uno strabismo da non uso.
Segni del fondo oculare
Sezione intitolata “Segni del fondo oculare”Al fondo oculare si osserva un difetto della papilla ottica e della retina e coroide inferiori, principalmente in basso. Sono presenti anomalie del decorso vascolare; l’arteria centrale della retina si divide posteriormente alla papilla, per cui molte arterie retiniche sembrano emergere dalla papilla. Il bordo superiore della papilla è spesso conservato, e anche quando l’intera papilla è depressa, la depressione è tipicamente più marcata in basso che in alto.
La regione papillare è depressa, la papilla è assente o parzialmente difettosa, e la coroide circostante, l’epitelio pigmentato retinico (EPR) e la sclera sono assottigliati. Al di sotto della depressione papillare, si osservano atrofia corioretinica e aspetto tigrato dovuti a una chiusura incompleta della fessura embrionale.
Classificazione
Sezione intitolata “Classificazione”Il coloboma della papilla ottica è classificato come segue in base all’estensione:
- Coloboma papillare isolato: dovuto a una chiusura incompleta localizzata della parte posteriore della fessura ottica.
- Coloboma corioretinico associato: indica una chiusura incompleta più estesa della fessura ottica.
- Tipo combinato coloboma irideo e ciliare : forma estesa continua dall’estremità anteriore a quella posteriore.
- Coloboma di Fuchs : forma lieve, che mostra una lesione atrofica simile a un cono al di sotto della papilla. La vista è spesso relativamente preservata.
Complicanze
Sezione intitolata “Complicanze”Complicanze oculari
Sezione intitolata “Complicanze oculari”- Spesso associato a coloboma irideo e coloboma coroideale.
- In caso di coloboma coroideale associato con ampia area di depressione, può verificarsi microftalmia.
- Distacco sieroso di retina: può verificarsi anche con coloboma isolato del disco ottico.
- Distacco regmatogeno di retina: può verificarsi secondariamente in casi di coloboma coroideale complesso.
- Ipotonia da filtrazione transclerale: sono stati riportati casi di fuoriuscita di umore acqueo attraverso il difetto sclerale 7).
3. Cause, epidemiologia e fattori di rischio
Sezione intitolata “3. Cause, epidemiologia e fattori di rischio”Epidemiologia
Sezione intitolata “Epidemiologia”La prevalenza è riportata tra 3 e 8/100.000. I casi unilaterali e bilaterali sono ugualmente frequenti, per lo più sporadici, ma spesso con storia familiare. Sono stati riportati vari modelli di ereditarietà: autosomico dominante, autosomico recessivo e legato all’X 4).
Il tasso complessivo di diagnosi genetica per i colobomi è inferiore al 30% 5). Molti casi sporadici senza mutazioni identificate suggeriscono il coinvolgimento di fattori ambientali e di più geni modificatori.
Meccanismo di sviluppo
Sezione intitolata “Meccanismo di sviluppo”La fessura embrionale (fessura della coppa ottica) si forma ventralmente durante la formazione della coppa ottica dal neuroectoderma alla 4a settimana di gestazione. Si completa alla 5a settimana e inizia a chiudersi dalla 6a settimana. La chiusura progredisce dalla regione equatoriale in avanti (verso l’iride) e indietro (verso il nervo ottico) e si completa alla 7a settimana. Un difetto localizzato di chiusura posteriore provoca un coloboma del nervo ottico.
Geni correlati
Sezione intitolata “Geni correlati”| Gene | Malattia associata | Note |
|---|---|---|
| PAX2 | Sindrome del coloboma renale (renal coloboma syndrome) | Coinvolto nella determinazione ventrale dell’occhio e nella chiusura della fessura embrionale1) |
| CHD7 | Sindrome CHARGE | Cromosoma 8 (8q12.2), malattia rara designata |
| FZD5 | Coloboma sindromico + microcornea | Recettore della via di segnalazione Wnt2) |
Complicanze sistemiche
Sezione intitolata “Complicanze sistemiche”Il coloboma del disco ottico può associarsi alle seguenti sindromi sistemiche.
- Sindrome CHARGE : sindrome malformativa multiorgano che prende le iniziali di coloboma (C), cardiopatia (H), atresia delle coane (A), ritardo di crescita (R), ipoplasia genitale (G) e anomalie dell’orecchio esterno (E). Il gene CHD7 è il gene responsabile; è riconosciuta come malattia rara designata.
- Sindrome di Aicardi : associata ad agenesia del corpo calloso, epilessia e ritardo dello sviluppo psicomotorio. Più frequente nelle bambine. Il coloboma si presenta spesso come multiple lacune nella coroide e nella retina.
- Sindrome coloboma renale : causata da mutazione del gene PAX2. Associata ad anomalie della formazione delle vie urinarie renali. È necessario un follow-up a lungo termine della funzione renale. La mutazione frameshift c.76delG di PAX2 è stata identificata in famiglie con glomerulosclerosi segmentaria e focale (FSGS), e lo spettro fenotipico è più ampio di quanto si pensasse in precedenza 1).
Può associarsi a sindromi sistemiche come la sindrome CHARGE (malformazioni multiorgano da mutazione di CHD7, malattia rara designata), la sindrome di Aicardi (agenesia del corpo calloso, epilessia, prevalenza femminile) e la sindrome coloboma renale (mutazione PAX2, anomalie delle vie urinarie renali). In caso di bilateralità o di reperti sistemici, si raccomandano consulenza pediatrica e consulenza genetica.
4. Diagnosi e metodi di esame
Sezione intitolata “4. Diagnosi e metodi di esame”Metodo diagnostico
Sezione intitolata “Metodo diagnostico”La diagnosi è possibile solo con l’oftalmoscopia. I punti chiave diagnostici sono una depressione bianca ben delimitata, prevalentemente nella parte inferiore del disco, e un’anomalia caratteristica del decorso vascolare (numerosi vasi che originano dal bordo o dall’interno della depressione). Per la diagnosi definitiva si utilizzano ecografia, RM, TC e tomografia a coerenza ottica (OCT).
Sebbene i casi sporadici siano frequenti, può esserci anche una storia familiare, pertanto è necessaria un’attenta raccolta dell’anamnesi familiare.
Per la ricerca di malformazioni intracraniche (come l’agenesia del corpo calloso) è necessaria una RM/TC cranica. Si effettua una consulenza pediatrica per verificare l’assenza di complicanze sistemiche come la sindrome CHARGE o la sindrome di Aicardi.
| Esame | Obiettivo |
|---|---|
| Esame del fondo oculare (dilatazione pupillare) | Valutazione della morfologia papillare, del decorso vascolare e del distacco di retina |
| OCT (tomografia a coerenza ottica) | Valutazione dettagliata della struttura della testa del nervo ottico e della macula |
| Ecografia (B-mode) | Ricerca di distacco di retina in caso di scarsa visibilità del fondo |
| Esame del campo visivo | Valutazione del pattern di difetto del campo visivo (difetto del campo visivo superiore, ecc.) |
| Risonanza magnetica cranica | Ricerca di complicanze del sistema nervoso centrale come agenesia del corpo calloso ed encefalocele |
| Ecografia renale | Screening per la sindrome del coloboma renale |
| Test genetico | Ricerca di mutazioni in PAX2, CHD7, ecc. (in casi bilaterali o sindromici) |
Diagnosi differenziale
Sezione intitolata “Diagnosi differenziale”| Diagnosi differenziale | Punti di differenziazione |
|---|---|
| Sindrome di morning glory | Proliferazione gliale al centro della papilla, vasi a decorso radiale. Nel coloboma, depressione inferiore senza proliferazione gliale |
| Stafiloma peripapillare | Ectasia posteriore della sclera intorno alla papilla. Il coloboma è un difetto della papilla stessa |
| PFV/PHPV papillare (persistenza dell’arteria ialoidea primitiva) | Con briglia vitreale e pieghe retiniche. Aspetto del fundus diverso dal coloboma |
| Megalopapilla | Diametro papillare aumentato ma morfologia quasi normale. Nessuna escavazione o anomalia vascolare 5) |
| Ipoplasia del nervo ottico | Papilla piccola (rapporto DM/DD ≥ 3,2). Il coloboma presenta papilla ingrandita ed escavata |
| Atrofia ottica glaucomatosa | Allargamento progressivo dell’escavazione e aumento della pressione intraoculare. Il coloboma è non progressivo con pressione normale |
5. Trattamento standard
Sezione intitolata “5. Trattamento standard”Il coloboma del disco ottico è un’anomalia strutturale congenita e non esiste un trattamento curativo. Il trattamento è principalmente sintomatico, in base alla presenza e al tipo di complicanze.
Osservazione
Sezione intitolata “Osservazione”Poiché si tratta di un’anomalia congenita non progressiva, in assenza di complicanze come il distacco sieroso della retina, si prosegue con l’osservazione regolare del fondo oculare. Nell’infanzia si raccomanda un esame del fondo oculare con pupilla dilatata ogni sei mesi-un anno.
Gestione del distacco sieroso della retina
Sezione intitolata “Gestione del distacco sieroso della retina”Non esiste un consenso sul trattamento del distacco sieroso della retina e sono stati riportati casi di risoluzione spontanea. A volte si osserva per alcuni mesi. Se non si osserva miglioramento dopo l’osservazione, si considera un intervento chirurgico.
Si ipotizza che l’anomalia strutturale dell’escavazione permetta al liquido vitreale di entrare nello spazio sottoretinico, ed è stata anche suggerita la possibilità di un afflusso di liquido cerebrospinale attraverso una comunicazione tra l’escavazione e lo spazio subaracnoideo.
Gestione del distacco regmatogeno della retina
Sezione intitolata “Gestione del distacco regmatogeno della retina”Per il distacco regmatogeno della retina si esegue vitrectomia e fotocoagulazione intorno all’escavazione. La prognosi visiva postoperatoria non è sempre favorevole.
Una tecnica di riattacco retinico con colla di fibrina è stata eseguita per il distacco di retina associato a coloboma, applicando colla di fibrina intorno alla lacerazione retinica al margine del coloboma per rafforzare l’adesione 3). In alcuni casi è stato ottenuto un miglioramento dell’acuità visiva finale a 20/50.
Trattamento dell’ambliopia
Sezione intitolata “Trattamento dell’ambliopia”In caso di scarsa acuità visiva, specialmente monolaterale, si esegue correzione refrattiva e terapia occlusiva (occlusione dell’occhio sano). L’intervento precoce nell’infanzia è importante. Tuttavia, in caso di riduzione della vista dovuta a un’anomalia strutturale del nervo ottico stesso, l’efficacia del trattamento dell’ambliopia è limitata.
Gestione sistemica
Sezione intitolata “Gestione sistemica”- Casi associati a sindrome CHARGE: necessaria gestione multidisciplinare con cardiochirurgia, ORL ed endocrinologia.
- Sindrome del coloboma renale: follow-up a lungo termine della funzione renale.
- Consulenza genetica: raccomandata in caso di bilateralità o sindrome.
Il distacco sieroso di retina può regredire spontaneamente, quindi si esegue prima un’osservazione di alcuni mesi. Per il distacco regmatogeno si esegue vitrectomia e fotocoagulazione intorno all’avvallamento. Recentemente sono stati riportati rinforzi dell’adesione con colla di fibrina. Tuttavia, la prognosi visiva postoperatoria non è sempre buona.
6. Fisiopatologia e meccanismi dettagliati
Sezione intitolata “6. Fisiopatologia e meccanismi dettagliati”Processo di chiusura della fessura embrionale
Sezione intitolata “Processo di chiusura della fessura embrionale”La coppa ottica si forma dal neuroectoderma alla 4a settimana di gestazione. Sulla superficie ventrale della coppa ottica si forma una fessura embrionale (fessura ottica), attraverso cui passa l’arteria ialoidea. Questa fessura si completa alla 5a settimana e inizia a chiudersi dalla 6a settimana. La chiusura inizia vicino all’equatore e progredisce anteriormente (verso l’iride) e posteriormente (verso il nervo ottico), completandosi alla 7a settimana. Un difetto localizzato di chiusura posteriore provoca il coloboma del nervo ottico.
Nel processo di chiusura è coinvolta la transizione epitelio-mesenchimale (EMT). Le cellule epiteliali della retina neurale al bordo della fessura embrionale degradano la membrana basale, acquisiscono caratteristiche mesenchimali e si fondono. Un’alterazione di questo processo causa il coloboma6).
Meccanismi molecolari
Sezione intitolata “Meccanismi molecolari”Il gene PAX2 è coinvolto nella determinazione ventrale dell’occhio e si ritiene che partecipi alla chiusura della fessura embrionale. Le mutazioni di PAX2 causano la sindrome del coloboma renale. Una mutazione frameshift c.76delG di PAX2 è stata identificata in una famiglia con FSGS, e il fenotipo della sindrome del coloboma renale è più ampio di quanto si pensasse in precedenza1).
Il gene FZD5 codifica un recettore della via di segnalazione Wnt. Le mutazioni ipofunzionali compromettono l’attivazione ligando-dipendente della segnalazione Wnt, causando un difetto di chiusura della fessura embrionale e microcornea. La modalità di trasmissione è recessiva2).
Il gene CHD7 codifica un fattore di rimodellamento della cromatina ed è coinvolto nella differenziazione e migrazione delle cellule della cresta neurale. Le mutazioni causano la sindrome CHARGE.
Meccanismo del distacco sieroso di retina
Sezione intitolata “Meccanismo del distacco sieroso di retina”Si ipotizza che, a causa di un’anomalia strutturale dell’escavazione, il liquido vitreale fluisca nello spazio sottoretinico. È stata anche suggerita la possibilità di un flusso di liquido cerebrospinale attraverso una comunicazione tra l’escavazione e lo spazio subaracnoideo, considerato uno dei motivi per cui il distacco sieroso della retina è difficile da trattare.
Prognosi e decorso
Sezione intitolata “Prognosi e decorso”Il coloboma del disco ottico è un’anomalia congenita fissa e non progressiva. La complicanza del distacco di retina è il principale fattore che peggiora la prognosi visiva. Anche dopo vitrectomia per distacco di retina regmatogeno, la prognosi visiva è spesso sfavorevole.
7. Ricerche recenti e prospettive future
Sezione intitolata “7. Ricerche recenti e prospettive future”Ampliamento dello spettro fenotipico delle mutazioni PAX2
Sezione intitolata “Ampliamento dello spettro fenotipico delle mutazioni PAX2”Hu et al. (2024) hanno riportato l’identificazione di una mutazione frameshift c.76delG in PAX2 in una famiglia con glomerulosclerosi segmentaria focale (FSGS) 1). Il fenotipo della sindrome del coloboma renale è più ampio di quanto si pensasse in precedenza, e l’importanza dello screening della funzione renale nei pazienti con coloboma del disco ottico è stata nuovamente sottolineata. Si suggerisce di ampliare le indicazioni per la valutazione della funzione renale nei pazienti con coloboma.
FZD5 e anomalie della segnalazione Wnt
Sezione intitolata “FZD5 e anomalie della segnalazione Wnt”Cortes-Gonzalez et al. (2024) hanno riportato che una mutazione missenso omozigote (p.M160V) in FZD5 causa coloboma oculare sintomatico e microcornea 2). L’analisi funzionale ha confermato che l’attivazione ligando-dipendente della via di segnalazione Wnt è compromessa con un modello di ereditarietà recessiva. Il tasso di diagnosi genetica del coloboma è inferiore al 30% e l’identificazione di nuovi geni causali dovrebbe contribuire a migliorare la diagnosi.
Nuove tecniche chirurgiche per il distacco di retina
Sezione intitolata “Nuove tecniche chirurgiche per il distacco di retina”Jain et al. (2024) hanno riportato un miglioramento dell’acuità visiva finale a 20/50 dopo retinopessia con colla di fibrina per distacco di retina associato a coloboma 3). La tecnica di applicare colla di fibrina attorno alla lacerazione retinica al bordo del coloboma per rafforzare l’adesione è considerata un’opzione aggiuntiva alla vitrectomia convenzionale con fotocoagulazione. L’accumulo di casi e la verifica dei risultati a lungo termine sono compiti futuri.
8. Riferimenti
Sezione intitolata “8. Riferimenti”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.