La sindrome di Aicardi è una malattia congenita rara descritta per la prima volta nel 1967 dal neurologo francese Jean Aicardi. Si presume sia legata all’X dominante, colpendo quasi esclusivamente le bambine. Nei maschi è letale in stato emizigote, con solo pochi casi riportati in maschi con cariotipo XXY (sindrome di Klinefelter).
L’incidenza è stimata in circa 1:110.000 nati, con circa 4.000 persone affette nel mondo 1). Tutti i casi sono mutazioni de novo, senza trasmissione da genitore a figlio, e il rischio di ricorrenza tra fratelli è inferiore all’1% 1).
La triade classica comprende i seguenti tre elementi 1):
Spasmi infantili: esordio intorno ai 3-4 mesi di età.
Lacune corioretiniche (chorioretinal lacunae) : lesioni rotondeggianti bilaterali del fondo oculare. Reperto specifico di questa malattia.
Agenesia del corpo calloso (agenesis of the corpus callosum) : assenza parziale o completa.
La prognosi è infausta. L’età media di sopravvivenza è di 18 anni e la probabilità di sopravvivere fino a 27 anni è riportata come 0,62% 1).
QLa sindrome di Aicardi si manifesta anche nei maschi?
A
Questa malattia si manifesta quasi esclusivamente nelle femmine. Si presume una trasmissione dominante legata all’X, poiché sarebbe letale nei maschi emizigoti. Tuttavia, esistono alcuni casi riportati in tutto il mondo in maschi con cariotipo XXY (sindrome di Klinefelter).
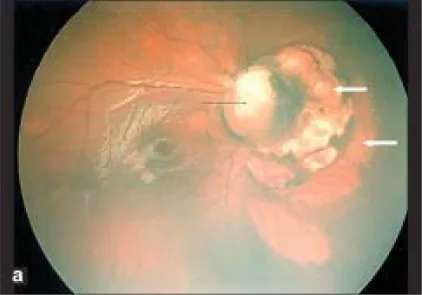
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Foto Retcam dell’occhio destro mostra coloboma del disco ottico (freccia nera) e aree pallide a forma di cupola con bordi netti in sede nasale rispetto al disco ottico, suggestive di lacune corioretiniche (frecce bianche).
Il sintomo iniziale di questa malattia sono solitamente gli spasmi infantili, che compaiono intorno ai 3-4 mesi di vita. L’epilessia spesso evolve in farmacoresistenza e si accompagna a vari tipi di crisi.
Crisi epilettiche : iniziano con spasmi infantili e progrediscono verso la farmacoresistenza. In un caso, a 4 mesi sono comparse convulsioni generalizzate 3-4 volte al giorno (della durata di 20-25 minuti ciascuna) 1). In un altro caso, sono state osservate frequenti crisi di ammiccamento a partire da 1 mese di età 2).
Ritardo psicomotorio : accompagnato da grave disabilità intellettiva, spesso con difficoltà nella deambulazione indipendente e nell’acquisizione del linguaggio.
Disfunzione gastrointestinale : sintomi digestivi come la stitichezza sono presenti in oltre il 90% dei casi 1).
Deficit visivo : causato da lesioni del fondo oculare, agenesia del corpo calloso e malformazioni corticali.
Tra i reperti oftalmologici di questa malattia, le lacune corioretiniche sono considerate patognomoniche.
Lacune corioretiniche
Distribuzione : bilaterale. Dense intorno alla papilla e al polo posteriore, ma si estendono anche alla periferia.
Aspetto : lesioni rotonde o ovali, di colore giallo-biancastro o rosa. Prevalenza 70–90%1).
Istologia : difetto dell’epitelio pigmentato retinico (RPE) che si estende dallo strato coroideo alla sclera nuda2).
Decorso : dimensioni e numero possono aumentare nel tempo dopo l’intervento chirurgico2).
Altri reperti oculari
Coloboma del nervo ottico : associato in circa il 44% dei casi.
Microftalmia : osservata in circa il 20% dei casi.
Retina periferica avascolare : può formarsi una zona avascolare a 360 gradi2).
Distacco di retina tractionale (TRD) : può verificarsi in associazione con tessuto peduncolare (stalk tissue)2).
In report di casi è stato documentato un esempio con emorragia preretinica e zona avascolare periferica a 360 gradi nell’occhio destro, e tessuto peduncolare (peduncolo fibrovascolare) con distacco di retina tractionale nell’occhio sinistro2). Può anche essere causa di deficit visivo corticale (CVI)3).
Agenesia del corpo calloso : assenza parziale o completa presente in tutti i casi1). È stata riportata anche la disgenesia (assottigliamento) del corpo calloso come variante2).
Malformazione della corteccia cerebrale: polimicrogiria (tipo 2 della classificazione di Barkovich), noduli di sostanza grigia periventricolari e cisti multiloculari sono visibili alla RM1).
EEG: mostra un pattern caratteristico con ritmi polimorfi ad alto voltaggio e scariche di punte-onda multifocali1).
Anomalie scheletriche: nel 40-60% si riscontrano fusioni vertebrali toraciche (T9-T10) o vertebre a farfalla (T8)1).
QLe lacune corioretiniche cambiano nel tempo?
A
Possono progredire. È stato riportato che dopo interventi chirurgici oculari compaiono nuove lacune, o che le loro dimensioni e numero aumentano nel tempo2). È importante un follow-up regolare con esame del fondo oculare.
La sindrome di Aicardi è considerata a ereditarietà X-linked dominante, ma il gene responsabile non è stato ancora identificato1). Tutti i casi sono mutazioni de novo e non si osserva generalmente ricorrenza familiare. Il rischio di ricorrenza per i fratelli è inferiore all’1% e si raccomanda di pianificare una successiva gravidanza dopo consulenza genetica1).
In un recente caso clinico, è stata rilevata una mutazione del gene TREX1 (c.292_293insA, p.(Cys99Metfs)) in un paziente2). TREX1 si trova sul cromosoma 3, il che contraddice in parte l’ipotesi X-linked, e la localizzazione del gene responsabile è ancora dibattuta.
Modalità di trasmissione: X-linked dominante (presunta). I maschi emizigoti sono letali in utero.
Natura delle mutazioni: tutte de novo. Non c’è trasmissione ereditaria (da genitore a figlio).
Rischio di ricorrenza: inferiore all’1% per i fratelli1).
La diagnosi della sindrome di Aicardi si basa principalmente sulla diagnosi clinica, poiché il gene responsabile non è stato identificato. La conferma della seguente triade classica è al centro della diagnosi1).
Spasmi infantili (infantile spasms)
Lacune corioretiniche (chorioretinal lacunae)
Agenesia del corpo calloso (agenesis of the corpus callosum)
Anche se sono presenti solo due dei tre segni, la diagnosi può essere posta secondo i criteri diagnostici estesi stabiliti nel 1999.
La composizione dei criteri diagnostici estesi è mostrata di seguito.
RM (cranio) : Conferma dell’agenesia del corpo calloso. Valutazione di polimicrogiria, eterotopia della sostanza grigia, dilatazione ventricolare laterale, cisti striatali talamiche bilaterali, ipoplasia ippocampale, ecc.1)2)
EEG (elettroencefalogramma) : Conferma di un ritmo polimorfo ad alto voltaggio e di un pattern di scariche punta-onda multifocali.1)
Esame del fondo oculare: conferma della presenza di lacune corioretiniche. L’angiografia con fluoresceina (FA) è utile per valutare le aree avascolari 2).
Radiografia del torace (AP): conferma di anomalie scheletriche (vertebre fuse, vertebre a farfalla) 1).
Collaborazione multidisciplinare: la diagnosi richiede la collaborazione dei reparti di neurologia, oftalmologia, ortopedia e genetica 1).
QIl test genetico può confermare la diagnosi?
A
Attualmente non è stato identificato alcun gene causale per una diagnosi definitiva, quindi il solo test genetico non può confermare la diagnosi 1). La diagnosi clinica si basa sulla combinazione di sintomi clinici, reperti del fondo oculare e reperti di imaging. I progressi nell’analisi genomica completa fanno sperare nell’identificazione del gene causale in futuro.
Non esiste una terapia curativa. Gli obiettivi del trattamento si basano su tre pilastri: controllo dell’epilessia, gestione delle complicanze oftalmiche e supporto allo sviluppo attraverso la riabilitazione.
Gestione dell'epilessia
Farmaci di prima linea: fenitoina, levetiracetam, clobazam, ecc. 1).
Casi refrattari: possono essere tentati cannabidiolo (CBD), dieta chetogenica, callosotomia, stimolazione del nervo vago 1).
Intervento oftalmico
Fotocoagulazione laser: eseguita sulla retina periferica avascolare per prevenire la progressione della retinopatia proliferante 2).
Vitrectomia: vitrectomia 23G per distacco di retina da trazione (TRD) 2).
Riabilitazione
Inizio precoce: iniziare fisioterapia, terapia occupazionale, logopedia e terapia visiva subito dopo la diagnosi 1).
Cure per ipovisione: fornire ausili visivi e adattamenti ambientali per i deficit visivi.
Vengono presentati esempi di interventi chirurgici per complicanze oftalmologiche riportati in letteratura.
Occhio
Reperti
Trattamento
Occhio destro
Emorragia preretinica, zona avascolare a 360°
Fotocoagulazione laser
Occhio sinistro
Tessuto peduncolato, TRD
Vitrectomia 23G
Dopo la vitrectomia è stato confermato il recupero della crescita della lunghezza assiale: la lunghezza assiale dell’occhio sinistro, che era di 17,45 mm a 1 mese di vita, è cresciuta fino a 24,41 mm a 26 mesi 2). Inoltre, sono stati riportati casi in cui dopo l’intervento sono diventate visibili nuove lacune corioretiniche, contribuendo alla conferma della diagnosi 2).
QÈ possibile un trattamento chirurgico oculistico?
A
Sì, è possibile. La fotocoagulazione laser della retina avascolare periferica e la vitrectomia 23G per il distacco di retina tractionale possono essere efficaci in alcuni casi 2). Ci sono anche segnalazioni di crescita oculare accelerata dopo l’intervento. Tuttavia, il numero di casi è ridotto ed è necessaria una gestione attenta in una struttura specializzata.
Si ritiene che una mutazione de novo sul cromosoma X sia la causa di questa malattia. Il modello di inattivazione del cromosoma X (lionizzazione) porterebbe a diversità fenotipica per la stessa mutazione. Nei maschi, la mutazione in stato emizigote è letale durante il periodo fetale, e solo i maschi con cariotipo XXY possono sopravvivere.
Recentemente, in un caso diagnosticato con questa malattia è stata rilevata una mutazione del gene TREX1 (c.292_293insA, p.(Cys99Metfs)) 2). Poiché TREX1 si trova sul cromosoma 3, ciò potrebbe contraddire l’ipotesi di un legame con l’X, suggerendo la possibilità di geni causali non legati all’X.
Le caratteristiche istologiche delle lacune corioretiniche includono un difetto dell’epitelio pigmentato retinico (RPE) che si estende dalla coriocapillare alla sclera nuda 2). Nelle aree di difetto, l’RPE e la coriocapillare sono assenti e si osserva displasia della retina neurosensoriale indifferenziata.
Si ritiene che i residui del sistema vascolare fetale persistente (persistent fetal vasculature) siano coinvolti nella formazione del tessuto fibrovascolare (stalk tissue) e della retina avascolare periferica 2). Questi vasi anomali causano il distacco di retina tractionale.
Il deficit visivo corticale (CVI) si verifica come una riduzione acquisita della funzione visiva dovuta ad anomalie strutturali cerebrali come polimicrogiria, eterotopia della sostanza grigia e agenesia del corpo calloso 3).
Le malformazioni cerebrali (polimicrogiria, agenesia del corpo calloso) sono basate su un disturbo della migrazione neuronale durante il periodo fetale. Il meccanismo molecolare di questo disturbo della migrazione rimane sconosciuto e sono attese future ricerche insieme all’identificazione del gene causale.
7. Ricerche recenti e prospettive future (rapporti in fase di ricerca)
Il gene responsabile rimane non identificato, ma la rilevazione di mutazioni del gene TREX1 2) è un nuovo indizio che suggerisce un possibile coinvolgimento di mutazioni non legate all’X. La diffusione dell’analisi genomica completa (WES/WGS) dovrebbe accelerare l’identificazione del gene responsabile. La verifica delle ipotesi di legame all’X e di non legame all’X è una sfida futura importante.
Riconoscimento delle forme varianti (atipiche) della sindrome di Aicardi
Si stanno accumulando segnalazioni di forme varianti che mostrano non un’assenza completa del corpo calloso, ma solo un assottigliamento (disgenesia) 2). Anche nei casi che non soddisfano tutti e tre i criteri, un esame dettagliato del fondo oculare e l’applicazione di criteri diagnostici estesi potrebbero migliorare l’accuratezza diagnostica.
Kang et al. (2022) hanno riportato l’esecuzione di fotocoagulazione laser e vitrectomia 23G in un caso di vitreoretinopatia bilaterale nella sindrome di Aicardi, contribuendo al mantenimento della crescita oculare e alla protezione della funzione visiva 2). Hanno anche registrato che dopo l’intervento sono diventate visibili nuove lacune corioretiniche, dimostrando che la chirurgia oftalmologica può contribuire anche alla conferma diagnostica.
Si riconosce sempre più l’importanza di uno screening precoce del fondo oculare e di un intervento rapido per proteggere la funzione retinica 2). Il laser sulle aree avascolari periferiche potrebbe prevenire la progressione della retinopatia proliferativa, e si auspica un accumulo di casi in futuro.
La ricerca sull’applicazione del cannabidiolo (CBD) e della dieta chetogenica per il controllo dell’epilessia sta progredendo 1). Si prevede l’istituzione di nuove opzioni terapeutiche per l’epilessia refrattaria.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.