A síndrome de Aicardi (Aicardi syndrome) é uma doença congênita rara descrita pela primeira vez em 1967 pelo neurologista francês Jean Aicardi. Acredita-se ser de herança dominante ligada ao X, e quase todos os pacientes são do sexo feminino. Em meninos, é letal em hemizigose, portanto apenas alguns casos foram relatados em meninos com cariótipo XXY (síndrome de Klinefelter).
A incidência estimada é de cerca de 1:110.000 nascimentos, e o número de afetados em todo o mundo é de aproximadamente 4.000 pessoas 1). Todos os casos são mutações de novo, sem transmissão de pais para filhos, e o risco de recorrência em irmãos é inferior a 1% 1).
A seguinte tríade clássica é conhecida 1):
Espasmos infantis (infantile spasms): início por volta dos 3-4 meses de idade.
Lacunas coriorretinianas (chorioretinal lacunae): Lesões fundoscópicas bilaterais e arredondadas. Achado específico desta doença.
Agenesia do corpo caloso (agenesis of the corpus callosum): Ausência parcial ou completa.
O prognóstico é ruim. A idade média de sobrevida é de 18 anos, e a probabilidade de sobreviver até os 27 anos é relatada como 0,62% 1).
QA síndrome de Aicardi também ocorre em meninos?
A
Esta doença ocorre quase exclusivamente em meninas. Supõe-se ser herança dominante ligada ao X, pois é letal em meninos hemizigotos. No entanto, existem alguns relatos de casos no mundo em meninos com cariótipo XXY (síndrome de Klinefelter).
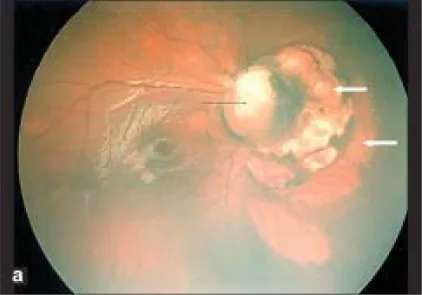
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Foto Retcam do olho direito mostra coloboma do disco óptico (seta preta) e áreas pálidas em forma de cúpula com bordas nítidas nasais ao disco óptico sugestivas de lacunas coriorretinianas (setas brancas).
O sintoma inicial desta doença é geralmente a epilepsia infantil (espasmos infantis) que aparece por volta dos 3-4 meses de idade. A epilepsia frequentemente se torna refratária a medicamentos e é acompanhada por vários tipos de crises.
Crises epilépticas: Iniciam-se com espasmos infantis e progridem para refratariedade medicamentosa. Em um caso, convulsões tônico-clônicas generalizadas ocorreram 3-4 vezes ao dia (cada uma durando 20-25 minutos) aos 4 meses de idade 1). Em outro caso, crises de piscar frequentes foram observadas desde 1 mês de idade 2).
Atraso do desenvolvimento psicomotor: Acompanhado de deficiência intelectual grave, muitas vezes é difícil alcançar mobilidade independente ou aquisição de linguagem.
Disfunção gastrointestinal: Sintomas gastrointestinais como constipação estão presentes em mais de 90% dos casos 1).
Deficiência visual: A deficiência visual ocorre devido a lesões fundoscópicas, agenesia do corpo caloso e malformações corticais.
Dentre os achados oftalmológicos desta doença, as lacunas coriorretinianas são consideradas um achado patognomônico.
Lacunas Coriorretinianas
Distribuição: Bilateral. Concentram-se ao redor do disco óptico e polo posterior, mas também se estendem à periferia.
Aparência: Lesões amarelo-esbranquiçadas a rosadas, redondas a ovais. Prevalência de 70-90%1).
Histologia: Defeito do epitélio pigmentar da retina (EPR) que se estende da camada coroidal até a camada escleral nua2).
Evolução: Tamanho e número podem aumentar ao longo do tempo após a cirurgia2).
Outros Achados Oculares
Coloboma do nervo óptico: Ocorre em cerca de 44% dos casos.
Microftalmia: Observada em cerca de 20% dos casos.
Retina avascular periférica: Pode formar-se uma zona avascular de 360 graus2).
Descolamento tracional da retina (DTR): Pode ocorrer associado a tecido pediculado (stalk tissue)2).
Em relatos de caso, foi registrado um exemplo com hemorragia pré-retiniana e zona avascular periférica de 360 graus no olho direito, e tecido pediculado (stalk tissue) com descolamento tracional da retina no olho esquerdo2). Também pode causar deficiência visual cortical (cortical visual impairment; CVI)3).
Agenesia do corpo caloso: Defeito parcial ou completo presente em todos os casos1). Disgenesia do corpo caloso também foi relatada como variante2).
Malformação do córtex cerebral: Polimicrogiria (tipo 2 de Barkovich), nódulos de substância cinzenta periventriculares e cistos multiloculados são confirmados por RM 1).
Achados de EEG: Mostra um padrão característico de ritmo polimórfico de alta voltagem com pontas multifocais e descargas de ondas 1).
Anomalias esqueléticas: Em 40-60% dos casos, observa-se fusão vertebral torácica (T9-T10), vértebra em borboleta (T8), entre outras 1).
QAs lacunas coriorretinianas mudam com o tempo?
A
Podem ser progressivas. Foi relatado que novas lacunas podem se tornar visíveis após intervenção cirúrgica ocular, ou que o tamanho e o número aumentam ao longo do tempo 2). O acompanhamento regular com exame de fundo de olho é importante.
A síndrome de Aicardi é presumida como uma herança dominante ligada ao X, mas o gene causador ainda não foi identificado 1). Todos os casos são mutações de novo e, basicamente, não há ocorrência familiar. O risco de recorrência para irmãos é inferior a 1%, e recomenda-se planejar o próximo filho após aconselhamento genético1).
Em relatos de casos recentes, uma mutação no gene TREX1 (c.292_293insA, p.(Cys99Metfs)) foi detectada em um caso 2). O TREX1 está localizado no cromossomo 3, o que contradiz parcialmente a hipótese de ligação ao X, e o debate sobre a localização do gene causador continua.
Padrão de herança: Dominante ligado ao X (presumido). Homens hemizigotos são letais no período fetal.
Natureza da mutação: Todas são mutações de novo. Não há herança (transmissão de pais para filhos).
Como o gene causador ainda não foi identificado, o diagnóstico da síndrome de Aicardi é baseado principalmente no diagnóstico clínico. A confirmação da tríade clássica a seguir é o centro do diagnóstico 1).
Espasmos infantis (infantile spasms)
Lacunas coriorretinianas (chorioretinal lacunae)
Agenesia do corpo caloso (agenesis of the corpus callosum)
Mesmo que apenas dois dos três critérios sejam atendidos, o diagnóstico pode ser feito usando os critérios diagnósticos expandidos estabelecidos em 1999.
Abaixo estão os componentes dos critérios diagnósticos expandidos.
Classificação
Itens principais
Características principais
Coloboma do nervo óptico, malformações corticais, heterotopia da substância cinzenta, cistos intracranianos, papiloma do plexo coroide
RM (cabeça): Confirmação de agenesia do corpo caloso. Avaliação de polimicrogiria, heterotopia da substância cinzenta, dilatação ventricular lateral, cistos talâmicos bilaterais, hipoplasia hipocampal, etc1)2).
EEG (eletroencefalograma): Confirmação do padrão de ritmo polimórfico de alta voltagem e ondas e pontas multifocais1).
Exame de fundo de olho: Confirmação da presença de lacunas coriorretinianas. A angiografia fluoresceínica (FA) é útil para avaliar áreas avasculares 2).
Raio-X da coluna torácica (AP): Confirmação de anormalidades esqueléticas (fusão vertebral, vértebra em borboleta) 1).
Colaboração multidisciplinar: Neurologia, oftalmologia, ortopedia e genética devem colaborar no diagnóstico 1).
QO teste genético pode confirmar o diagnóstico?
A
Atualmente, nenhum gene causador foi identificado para confirmar o diagnóstico, portanto o diagnóstico não pode ser estabelecido apenas por teste genético 1). O diagnóstico clínico baseado na combinação de sintomas, achados de fundo de olho e imagem é o principal. Com o avanço da análise genômica abrangente, espera-se que o gene causador seja identificado no futuro.
Não existe tratamento curativo. O tratamento visa três pilares: controle das convulsões, manejo das complicações oftalmológicas e suporte ao desenvolvimento por meio de reabilitação.
Manejo da epilepsia
Medicamentos de primeira linha: Fenitoína, levetiracetam, clobazam, entre outros 1).
Casos refratários: Canabidiol (CBD), dieta cetogênica, calosotomia, estimulação do nervo vago podem ser tentados 1).
Intervenção oftalmológica
Fotocoagulação a laser: Realizada na retina avascular periférica para prevenir a progressão da retinopatia proliferativa 2).
Vitrectomia: Vitrectomia 23G é realizada para descolamento de retina tracional (TRD) 2).
Reabilitação
Início precoce: Iniciar fisioterapia, terapia ocupacional, fonoaudiologia e terapia visual imediatamente após o diagnóstico 1).
Cuidados para baixa visão: Fornecimento de dispositivos auxiliares e adaptações ambientais para deficiência visual.
Abaixo estão exemplos de intervenções cirúrgicas para complicações oftalmológicas.
Olho
Achados
Tratamento
Olho direito
Hemorragia pré-retiniana e zona avascular de 360 graus
Fotocoagulação a laser
Olho esquerdo
Tecido em haste e descolamento tracional da retina
Vitrectomia 23G
Após a vitrectomia, a recuperação do crescimento do comprimento axial foi confirmada; no olho esquerdo, o comprimento axial cresceu de 17,45 mm com 1 mês de idade para 24,41 mm com 26 meses2). Além disso, foram relatados casos em que lacunas coriorretinianas se tornaram visíveis após a cirurgia, auxiliando na confirmação do diagnóstico2).
QO tratamento cirúrgico oftalmológico é possível?
A
Sim, é possível. A fotocoagulação a laser da retina avascular periférica e a vitrectomia 23G para descolamento de retina tracional podem ser eficazes em alguns casos2). Há relatos de aceleração do crescimento ocular após a cirurgia. No entanto, o número de casos é pequeno, exigindo manejo cuidadoso em centros especializados.
Mutações de novo no cromossomo X são consideradas a causa desta doença. Acredita-se que o padrão de inativação do cromossomo X (lionização) cause diversidade fenotípica mesmo com a mesma mutação. Em homens, por ser hemizigoto, leva à letalidade embrionária, e apenas homens com cariótipo XXY podem sobreviver.
Recentemente, uma mutação no gene TREX1 (c.292_293insA, p.(Cys99Metfs)) foi detectada em um caso diagnosticado com esta doença2). Como o TREX1 está localizado no cromossomo 3, isso pode contradizer a hipótese ligada ao X, sugerindo a possibilidade de um gene causador não ligado ao X.
Como característica histológica das lacunas coriorretinianas, foi confirmado que o defeito do epitélio pigmentar da retina (EPR) se estende da lâmina coriocapilar até a esclera nua2). Não há epitélio pigmentar da retina nem lâmina coriocapilar no local do defeito, e observa-se displasia da retina neural indiferenciada.
Acredita-se que resquícios do sistema vascular fetal persistente (persistent fetal vasculature) estejam envolvidos na formação do pedículo fibrovascular (stalk tissue) e da retina avascular periférica2). Esses vasos anormais causam descolamento de retina tracional.
A deficiência visual cortical (CVI) é uma redução adquirida da função visual decorrente de anormalidades estruturais cerebrais como polimicrogiria, heterotopia de substância cinzenta e agenesia do corpo caloso3).
As malformações cerebrais (polimicrogiria, agenesia do corpo caloso) baseiam-se em um distúrbio da migração neuronal durante o período embrionário. O mecanismo molecular causador desse distúrbio de migração permanece desconhecido, e pesquisas futuras são aguardadas juntamente com a identificação do gene causador.
7. Pesquisas mais recentes e perspectivas futuras (relatos em fase de pesquisa)
O gene causador ainda não foi identificado, mas a detecção da mutação no gene TREX1 2) é uma nova pista que sugere a possível participação de mutações não ligadas ao X. Com a disseminação da análise genômica abrangente (WES e WGS), espera-se que a identificação do gene causador seja acelerada. A verificação das hipóteses ligada ao X e não ligada ao X é uma tarefa principal futura.
Reconhecimento da síndrome de Aicardi variante (atípica)
Relatos de variantes que não apresentam agenesia completa do corpo caloso, mas apenas afinamento (disgenesia), estão se acumulando 2). Mesmo em casos que não preenchem todos os três critérios, a aplicação de exame detalhado de fundo de olho e critérios diagnósticos expandidos pode melhorar a precisão diagnóstica.
Kang et al. (2022) relataram a realização de fotocoagulação a laser e vitrectomia 23G em um caso de vitreorretinopatia bilateral na síndrome de Aicardi, contribuindo para a manutenção do crescimento do globo ocular e proteção da função visual 2). Também foi registrado que lacunas coriorretinianas se tornaram visíveis após a cirurgia, indicando que a cirurgia ocular pode contribuir para a confirmação do diagnóstico.
Há um reconhecimento crescente de que a triagem precoce do fundo de olho e a intervenção rápida são importantes para proteger a função da retina2). O laser em áreas avasculares periféricas tem potencial para prevenir a progressão da retinopatia proliferativa, e espera-se o acúmulo de mais casos no futuro.
Pesquisas sobre o uso de canabidiol (CBD) e terapia dietética cetogênica para controle da epilepsia estão em andamento 1). Espera-se o estabelecimento de novas opções de tratamento para epilepsia refratária.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.