Le syndrome d’Aicardi est une maladie congénitale rare décrite pour la première fois en 1967 par le neurologue français Jean Aicardi. Il est supposé être lié à l’X dominant, touchant presque exclusivement les filles. Chez les garçons, il est létal à l’état hémizygote, seuls quelques cas étant rapportés chez des garçons avec caryotype XXY (syndrome de Klinefelter).
L’incidence est estimée à environ 1:110 000 naissances, avec environ 4 000 personnes atteintes dans le monde 1). Tous les cas sont des mutations de novo, sans transmission parent-enfant, et le risque de récidive chez les frères et sœurs est inférieur à 1 % 1).
La triade classique comprend les trois éléments suivants 1) :
Spasmes infantiles : apparaissant vers l’âge de 3 à 4 mois.
Lacunes chorio-rétiniennes (chorioretinal lacunae) : lésions rondes bilatérales du fond d’œil. Signe spécifique de cette maladie.
Agénésie du corps calleux (agenesis of the corpus callosum) : absence partielle ou complète.
Le pronostic est sombre. L’âge moyen de survie est de 18 ans, et la probabilité de survivre jusqu’à 27 ans est rapportée à 0,62 % 1).
QLe syndrome d'Aicardi survient-il aussi chez les garçons ?
A
Cette maladie survient presque exclusivement chez les filles. On suppose une transmission dominante liée à l’X, car elle serait létale chez les garçons hémizygotes. Cependant, quelques cas ont été rapportés dans le monde chez des garçons présentant un caryotype XXY (syndrome de Klinefelter).
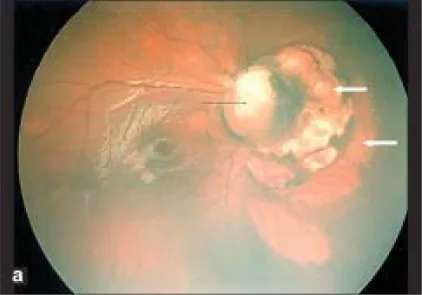
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Photo Retcam de l’œil droit montrant un colobome du disque optique (flèche noire) et des zones pâles en forme de dôme aux bords nets en nasal du disque optique, évocatrices de lacunes chorio-rétiniennes (flèches blanches).
Le symptôme initial de cette maladie est généralement une épilepsie en salves apparaissant vers l’âge de 3 à 4 mois. L’épilepsie évolue souvent vers une résistance aux médicaments et s’accompagne de divers types de crises.
Crises d’épilepsie : débutent par une épilepsie en salves et évoluent vers une résistance aux médicaments. Dans un cas, des convulsions généralisées sont apparues à 4 mois, 3 à 4 fois par jour (durée de 20 à 25 minutes chacune) 1). Dans un autre cas, des crises d’épilepsie fréquentes avec clignement des yeux ont été observées dès l’âge de 1 mois 2).
Retard psychomoteur : accompagné d’un handicap intellectuel sévère, rendant souvent difficile la mobilité indépendante et l’acquisition du langage.
Dysfonctionnement gastro-intestinal : des symptômes digestifs tels que la constipation sont présents dans plus de 90 % des cas 1).
Déficience visuelle : due aux lésions du fond d’œil, à l’agénésie du corps calleux et aux malformations corticales.
Des rapports de cas ont documenté un exemple avec une hémorragie prérétinienne et une zone avasculaire périphérique à 360 degrés dans l’œil droit, et un tissu pédiculaire (stalk fibrovasculaire) avec décollement de rétine tractionnel dans l’œil gauche2). Cela peut également être une cause de déficience visuelle corticale (CVI)3).
Agénésie du corps calleux : absence partielle ou complète présente dans tous les cas1). Une dysgénésie du corps calleux (amincissement) a également été rapportée comme variante2).
Malformation du cortex cérébral : polymicrogyrie (type 2 de la classification de Barkovich), nodules de substance grise périventriculaires et kystes multiloculaires sont observés à l’IRM1).
EEG : montre un motif caractéristique de rythmes polymorphes de haut voltage avec des pointes multifocales et des décharges d’ondes1).
Anomalies squelettiques : 40 à 60 % présentent une fusion des vertèbres thoraciques (T9-T10) ou des vertèbres en papillon (T8)1).
QLes lacunes choroïdorétiniennes changent-elles avec le temps ?
A
Elles peuvent progresser. Il a été rapporté que de nouvelles lacunes deviennent visibles après une intervention chirurgicale ophtalmique, ou que leur taille et leur nombre augmentent avec le temps2). Un suivi régulier par examen du fond d’œil est important.
Le syndrome d’Aicardi est supposé être lié à l’X dominant, mais le gène responsable n’a pas encore été identifié1). Tous les cas sont des mutations de novo, et une transmission familiale n’est généralement pas observée. Le risque de récidive pour les frères et sœurs est inférieur à 1 %, et il est recommandé de discuter de la planification d’une future grossesse après un conseil génétique1).
Dans un récent rapport de cas, une mutation du gène TREX1 (c.292_293insA, p.(Cys99Metfs)) a été détectée chez un patient2). TREX1 est situé sur le chromosome 3, ce qui contredit en partie l’hypothèse d’une liaison à l’X, et la localisation du gène responsable fait toujours débat.
Mode de transmission : lié à l’X dominant (supposé). Les hommes hémizygotes sont létaux in utero.
Nature des mutations : toutes de novo. Il n’y a pas de transmission héréditaire (de parent à enfant).
Risque de récidive : inférieur à 1 % pour les frères et sœurs1).
Le diagnostic du syndrome d’Aicardi repose principalement sur le diagnostic clinique, car le gène responsable n’est pas encore identifié. La confirmation de la triade classique suivante est au cœur du diagnostic1).
IRM (crânienne) : Confirmation de l’agénésie du corps calleux. Évaluation de la polymicrogyrie, de l’hétérotopie de la substance grise, de la dilatation ventriculaire latérale, des kystes striés thalamiques bilatéraux, de l’hypoplasie hippocampique, etc.1)2)
EEG (électroencéphalogramme) : Confirmation d’un rythme polymorphe de haut voltage et d’un motif de pointes-ondes multifocales et de décharges de pointes.1)
Examen du fond d’œil : confirmation de la présence de lacunes choroïdorétiniennes. L’angiographie à la fluorescéine (FA) est utile pour évaluer les zones avasculaires 2).
Radiographie thoracique (AP) : confirmation des anomalies squelettiques (vertèbres fusionnées, vertèbres en papillon) 1).
Collaboration multidisciplinaire : le diagnostic nécessite la collaboration des services de neurologie, d’ophtalmologie, d’orthopédie et de génétique 1).
QUn test génétique peut-il confirmer le diagnostic ?
A
À l’heure actuelle, aucun gène causal n’a été identifié pour un diagnostic définitif, donc un test génétique seul ne peut pas confirmer le diagnostic 1). Le diagnostic clinique repose sur la combinaison des symptômes cliniques, des résultats du fond d’œil et de l’imagerie. Les progrès de l’analyse génomique exhaustive devraient permettre d’identifier le gène causal à l’avenir.
Il n’existe pas de traitement curatif. Les objectifs du traitement reposent sur trois piliers : le contrôle de l’épilepsie, la prise en charge des complications ophtalmiques et le soutien au développement par la rééducation.
Gestion de l'épilepsie
Traitement de première intention : phénytoïne, lévétiracétam, clobazam, etc. 1).
Cas réfractaires : cannabidiol (CBD), régime cétogène, callosotomie, stimulation du nerf vague peuvent être tentés 1).
Intervention ophtalmique
Photocoagulation au laser : réalisée sur la rétine périphérique avasculaire pour prévenir la progression de la rétinopathie proliférante 2).
Voici des exemples de rapports d’interventions chirurgicales pour les complications ophtalmologiques.
Œil
Constatations
Traitement
Œil droit
Hémorragie prérétinienne, zone avasculaire à 360°
Photocoagulation au laser
Œil gauche
Tissu pédonculé, TRD
Vitrectomie 23G
Après la vitrectomie, une récupération de la croissance de la longueur axiale a été confirmée : la longueur axiale de l’œil gauche, qui était de 17,45 mm à 1 mois après la naissance, a atteint 24,41 mm à 26 mois 2). De plus, des cas ont été rapportés où des lacunes choroïdorétiniennes sont devenues visibles après l’opération, contribuant à la confirmation du diagnostic 2).
QUn traitement chirurgical ophtalmique est-il possible ?
A
Oui, c’est possible. La photocoagulation au laser de la rétine avasculaire périphérique et la vitrectomie 23G pour le décollement de rétine tractionnel peuvent être efficaces dans certains cas 2). Des rapports indiquent une croissance oculaire accélérée après la chirurgie. Cependant, le nombre de cas est faible et une prise en charge prudente dans un établissement spécialisé est nécessaire.
On pense qu’une mutation de novo sur le chromosome X est la cause de cette maladie. Le modèle d’inactivation du chromosome X (lyonisation) entraînerait une diversité phénotypique pour une même mutation. Chez les hommes, la mutation serait létale à l’état hémizygote pendant la période fœtale, seuls les hommes avec un caryotype XXY pouvant survivre.
Récemment, une mutation du gène TREX1 (c.292_293insA, p.(Cys99Metfs)) a été détectée chez un patient diagnostiqué avec cette maladie 2). Comme TREX1 est situé sur le chromosome 3, cela pourrait contredire l’hypothèse de liaison à l’X, suggérant la possibilité de gènes responsables non liés à l’X.
Les caractéristiques histologiques des lacunes choroïdorétiniennes comprennent une perte de l’épithélium pigmentaire rétinien (EPR) s’étendant de la choriocapillaire à la sclère nue 2). Dans les zones de perte, l’EPR et la choriocapillaire sont absents, et on observe une dysplasie de la rétine neurale indifférenciée.
La déficience visuelle corticale (CVI) résulte d’une diminution acquise de la fonction visuelle due à des anomalies structurelles cérébrales telles que la polymicrogyrie, l’hétérotopie de la substance grise et l’agénésie du corps calleux 3).
Les malformations cérébrales (polymicrogyrie, agénésie du corps calleux) sont basées sur un trouble de la migration neuronale pendant la période fœtale. Le mécanisme moléculaire à l’origine de ce trouble de la migration reste inconnu, et des recherches futures sont attendues parallèlement à l’identification du gène responsable.
7. Recherches récentes et perspectives d’avenir (rapports en phase de recherche)
Le gène responsable reste non identifié, mais la détection de mutations du gène TREX1 2) est un nouvel indice suggérant l’implication possible de mutations non liées à l’X. La généralisation de l’analyse génomique exhaustive (WES/WGS) devrait accélérer l’identification du gène responsable. La vérification des hypothèses de liaison à l’X et de non-liaison à l’X constitue un défi majeur pour l’avenir.
Reconnaissance des formes variantes (atypiques) du syndrome d’Aicardi
Des rapports de formes variantes montrant non pas une agénésie complète du corps calleux mais seulement une dysgénésie (amincissement) s’accumulent 2). Même dans les cas ne remplissant pas tous les trois critères, un examen détaillé du fond d’œil et l’application de critères diagnostiques élargis pourraient améliorer la précision du diagnostic.
Importance de l’intervention ophtalmologique précoce
Kang et al. (2022) ont rapporté avoir réalisé une photocoagulation au laser et une vitrectomie 23G dans un cas de vitréorétinopathie bilatérale du syndrome d’Aicardi, contribuant au maintien de la croissance oculaire et à la protection de la fonction visuelle 2). Ils ont également noté que des lacunes choroïdorétiniennes sont devenues visibles après l’opération, montrant que la chirurgie ophtalmologique peut également contribuer à la confirmation du diagnostic.
On reconnaît de plus en plus l’importance d’un dépistage précoce du fond d’œil et d’une intervention rapide pour protéger la fonction rétinienne 2). Le laser sur les zones avasculaires périphériques pourrait prévenir la progression de la rétinopathie proliférative, et une accumulation de cas est souhaitée à l’avenir.
La recherche sur l’application du cannabidiol (CBD) et du régime cétogène pour le contrôle de l’épilepsie progresse 1). On espère l’établissement de nouvelles options thérapeutiques pour l’épilepsie réfractaire.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.