Hội chứng Aicardi (Aicardi syndrome) là một bệnh bẩm sinh hiếm gặp được bác sĩ thần kinh người Pháp Jean Aicardi mô tả lần đầu tiên vào năm 1967. Bệnh được cho là di truyền trội liên kết X, và hầu như tất cả bệnh nhân đều là nữ. Ở nam giới, bệnh gây chết ở thể bán hợp tử, do đó chỉ có một số ít trường hợp được báo cáo ở nam giới có kiểu nhân XXY (hội chứng Klinefelter).
Tỷ lệ mắc ước tính khoảng 1:110.000 ca sinh, và số người mắc bệnh trên toàn thế giới khoảng 4.000 người 1). Tất cả các trường hợp đều là đột biến mới (de novo), không có trường hợp truyền từ cha mẹ sang con, và nguy cơ tái phát ở anh chị em ruột dưới 1% 1).
Bộ ba kinh điển sau đây được biết đến 1):
Co thắt trẻ sơ sinh (infantile spasms): khởi phát vào khoảng 3-4 tháng tuổi.
Khuyết hắc võng mạc (chorioretinal lacunae): Tổn thương đáy mắt hình tròn hai bên. Dấu hiệu đặc hiệu của bệnh này.
Bất sản thể chai (agenesis of the corpus callosum): Thiếu hụt một phần hoặc toàn bộ.
Tiên lượng xấu. Tuổi thọ trung bình là 18 tuổi, và xác suất sống đến 27 tuổi được báo cáo là 0,62% 1).
QHội chứng Aicardi có xảy ra ở bé trai không?
A
Bệnh này hầu như chỉ xảy ra ở bé gái. Được cho là di truyền trội liên kết X, vì ở bé trai thể dị hợp tử sẽ gây chết. Tuy nhiên, có một vài báo cáo ca bệnh trên thế giới ở bé trai có kiểu nhân XXY (hội chứng Klinefelter).
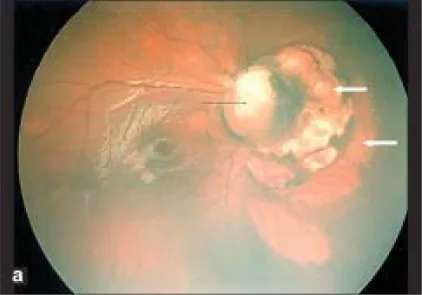
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Ảnh Retcam mắt phải cho thấy u nguyên bào thần kinh thị giác (mũi tên đen) và các vùng nhạt màu hình vòm có ranh giới rõ ở phía mũi của đĩa thị giác gợi ý khuyết hắc võng mạc (mũi tên trắng).
Triệu chứng khởi phát của bệnh này thường là động kinh giật cơ ở trẻ nhũ nhi (co thắt trẻ sơ sinh) xuất hiện vào khoảng 3-4 tháng tuổi. Động kinh thường trở nên kháng thuốc và kèm theo nhiều loại cơn khác nhau.
Cơn động kinh: Khởi phát bằng co thắt trẻ sơ sinh và tiến triển thành kháng thuốc. Trong một ca, co giật toàn thể xảy ra 3-4 lần/ngày (mỗi lần kéo dài 20-25 phút) lúc 4 tháng tuổi 1). Trong một ca khác, các cơn chớp mắt thường xuyên được quan sát từ 1 tháng tuổi 2).
Chậm phát triển tâm thần vận động: Kèm theo khuyết tật trí tuệ nặng, thường khó đạt được khả năng vận động độc lập hoặc tiếp thu ngôn ngữ.
Rối loạn chức năng tiêu hóa: Các triệu chứng tiêu hóa như táo bón có ở hơn 90% trường hợp 1).
Suy giảm thị lực: Suy giảm thị lực xảy ra do tổn thương đáy mắt, bất sản thể chai và dị dạng vỏ não.
Trong số các dấu hiệu nhãn khoa của bệnh này, các khuyết hổng hắc võng mạc (choroidal retinal lacunae) được coi là dấu hiệu bệnh lý đặc hiệu (pathognomonic).
Khuyết hổng hắc võng mạc
Phân bố: Hai bên. Tập trung quanh gai thị và cực sau, nhưng cũng lan ra vùng ngoại vi.
Hình dạng: Tổn thương hình tròn đến bầu dục, màu trắng-vàng đến hồng. Tỷ lệ mắc 70-90%1).
Nhãn cầu nhỏ (microphthalmia): Gặp ở khoảng 20% trường hợp.
Võng mạc vô mạch ngoại vi: Có thể hình thành vùng vô mạch 360 độ2).
Bong võng mạc co kéo (TRD): Có thể xảy ra cùng với mô cuống (stalk tissue)2).
Trong báo cáo ca bệnh, đã ghi nhận trường hợp có xuất huyết trước võng mạc và vùng vô mạch ngoại vi 360 độ ở mắt phải, và mô cuống (stalk tissue) kèm bong võng mạc co kéo ở mắt trái2). Ngoài ra, có thể gây suy giảm thị lực vỏ não (cortical visual impairment; CVI)3).
Bất sản thể chai: Khiếm khuyết một phần hoặc hoàn toàn có ở tất cả các trường hợp1). Loạn sản thể chai (dysgenesis) cũng được báo cáo như một biến thể2).
Dị dạng vỏ não: Đa hồi não nhỏ (polymicrogyria, phân loại Barkovich type 2), các nốt chất xám quanh não thất và nang nhiều ngăn được xác nhận trên MRI 1).
Kết quả EEG: Cho thấy mô hình đặc trưng của nhịp đa hình thái điện thế cao kèm theo các sóng nhọn đa ổ và phóng lực sóng 1).
Bất thường xương: 40-60% trường hợp có dính đốt sống ngực (T9-T10), đốt sống hình cánh bướm (T8) và các bất thường khác 1).
QCác khuyết hắc võng mạc có thay đổi theo thời gian không?
A
Có thể tiến triển. Đã có báo cáo về các khuyết mới xuất hiện sau can thiệp phẫu thuật mắt, hoặc kích thước và số lượng tăng dần theo thời gian 2). Việc theo dõi định kỳ bằng khám đáy mắt là rất quan trọng.
Hội chứng Aicardi được cho là di truyền trội liên kết nhiễm sắc thể X, nhưng gen gây bệnh hiện vẫn chưa được xác định 1). Tất cả các trường hợp đều là đột biến mới (de novo), và về cơ bản không thấy có trường hợp gia đình. Nguy cơ tái phát cho anh chị em ruột dưới 1%, và nên thảo luận về kế hoạch sinh con tiếp theo sau khi tư vấn di truyền1).
Trong các báo cáo ca bệnh gần đây, đột biến gen TREX1 (c.292_293insA, p.(Cys99Metfs)) đã được phát hiện trong một trường hợp 2). TREX1 nằm trên nhiễm sắc thể số 3, một phần mâu thuẫn với giả thuyết liên kết X, do đó vẫn còn tranh luận về vị trí của gen gây bệnh.
Kiểu di truyền: Trội liên kết X (giả định). Nam giới bán hợp tử gây chết trong giai đoạn bào thai.
Bản chất đột biến: Tất cả đều là đột biến mới (de novo). Không có tính di truyền (từ cha mẹ sang con).
Vì gen gây bệnh chưa được xác định, chẩn đoán hội chứng Aicardi chủ yếu dựa trên chẩn đoán lâm sàng. Việc xác nhận bộ ba kinh điển sau đây là trọng tâm của chẩn đoán 1).
Co thắt ở trẻ nhũ nhi (infantile spasms)
Khuyết hắc võng mạc (chorioretinal lacunae)
Bất sản thể chai (agenesis of the corpus callosum)
Ngay cả khi chỉ có hai trong ba triệu chứng, vẫn có thể chẩn đoán bằng tiêu chuẩn chẩn đoán mở rộng được thiết lập năm 1999.
Dưới đây là các thành phần của tiêu chuẩn chẩn đoán mở rộng.
MRI (đầu): Xác nhận bất sản thể chai. Đánh giá đa hồi nhỏ, lạc chỗ chất xám, giãn não thất bên, nang đồi thị hai bên, giảm sản hồi hải mã, v.v.1)2).
EEG (điện não đồ): Xác nhận mô hình nhịp đa dạng điện thế cao và sóng nhọn đa ổ1).
Khám đáy mắt: Xác nhận sự hiện diện của các lacuna hắc võng mạc. Chụp mạch huỳnh quang (FA) hữu ích để đánh giá vùng vô mạch 2).
X-quang cột sống ngực (AP): Xác nhận bất thường xương (dính đốt sống, đốt sống hình cánh bướm) 1).
Phối hợp đa chuyên khoa: Thần kinh, nhãn khoa, chỉnh hình và di truyền cần phối hợp trong chẩn đoán 1).
QXét nghiệm di truyền có thể chẩn đoán xác định không?
A
Hiện tại, chưa xác định được gen gây bệnh để chẩn đoán xác định, do đó không thể chẩn đoán chỉ dựa vào xét nghiệm di truyền 1). Chẩn đoán lâm sàng dựa trên tổng hợp triệu chứng, hình ảnh đáy mắt và hình ảnh học là chủ yếu. Với sự tiến bộ của phân tích hệ gen toàn diện, hy vọng sẽ xác định được gen gây bệnh trong tương lai.
Không có phương pháp điều trị triệt căn. Mục tiêu điều trị gồm ba trụ cột: kiểm soát cơn động kinh, xử lý biến chứng nhãn khoa và hỗ trợ phát triển thông qua phục hồi chức năng.
Quản lý động kinh
Thuốc hàng đầu: Phenytoin, levetiracetam, clobazam và các thuốc khác 1).
Trường hợp kháng trị: Có thể thử cannabidiol (CBD), chế độ ăn ketogenic, phẫu thuật cắt thể chai, kích thích thần kinh phế vị 1).
Can thiệp nhãn khoa
Quang đông laser: Thực hiện trên võng mạc vô mạch ngoại vi để ngăn ngừa tiến triển bệnh võng mạc tăng sinh 2).
Cắt dịch kính: Cắt dịch kính 23G được thực hiện cho bong võng mạc co kéo (TRD) 2).
Phục hồi chức năng
Bắt đầu sớm: Bắt đầu vật lý trị liệu, hoạt động trị liệu, ngôn ngữ trị liệu và thị giác trị liệu ngay sau khi chẩn đoán 1).
Chăm sóc thị lực kém: Cung cấp dụng cụ hỗ trợ và điều chỉnh môi trường cho người khiếm thị.
Sau cắt dịch kính, sự phục hồi tăng trưởng chiều dài trục nhãn cầu đã được xác nhận; ở mắt trái, chiều dài trục tăng từ 17,45 mm lúc 1 tháng tuổi lên 24,41 mm lúc 26 tháng tuổi2). Ngoài ra, đã có báo cáo về các trường hợp các lỗ khuyết hắc võng mạc mới xuất hiện sau phẫu thuật, giúp xác nhận chẩn đoán2).
QCó thể điều trị phẫu thuật mắt không?
A
Có thể. Quang đông laservõng mạc vô mạch ngoại vi và cắt dịch kính 23G cho bong võng mạc co kéo có thể hiệu quả trong một số trường hợp2). Có báo cáo về sự thúc đẩy tăng trưởng nhãn cầu sau phẫu thuật. Tuy nhiên, số ca ít, đòi hỏi xử trí thận trọng tại các cơ sở chuyên khoa.
Đột biến de novo trên nhiễm sắc thể X được cho là nguyên nhân của bệnh này. Mô hình bất hoạt nhiễm sắc thể X (lion hóa) được cho là gây ra sự đa dạng kiểu hình ngay cả với cùng một đột biến. Ở nam giới, do thể bán hợp tử, gây chết trong giai đoạn phôi thai, và chỉ nam giới có kiểu nhân XXY mới có thể sống sót.
Gần đây, đột biến gen TREX1 (c.292_293insA, p.(Cys99Metfs)) đã được phát hiện trong một trường hợp được chẩn đoán mắc bệnh này2). Vì TREX1 nằm trên nhiễm sắc thể 3, điều này có thể mâu thuẫn với giả thuyết liên kết X, cho thấy khả năng tồn tại gen gây bệnh không liên kết X.
Là đặc điểm mô học của lacuna hắc võng mạc, đã được xác nhận rằng khiếm khuyết biểu mô sắc tố võng mạc (RPE) kéo dài từ lớp mao mạch hắc mạc đến lớp củng mạc trần2). Không có biểu mô sắc tố võng mạc và lớp mao mạch hắc mạc tại vị trí khiếm khuyết, và quan sát thấy loạn sản võng mạc thần kinh chưa biệt hóa.
Phần còn lại của hệ thống mạch máu thai nhi tồn tại (persistent fetal vasculature) được cho là có liên quan đến sự hình thành cuống xơ mạch (stalk tissue) và võng mạc vô mạch ngoại vi2). Các mạch máu bất thường này gây ra bong võng mạc co kéo.
Rối loạn thị giác vỏ não (CVI) là sự suy giảm chức năng thị giác mắc phải do các bất thường cấu trúc não như đa hồi nhỏ (polymicrogyria), chất xám lạc chỗ (heterotopic gray matter) và bất sản thể chai (agenesis of corpus callosum)3).
Dị dạng não (đa hồi nhỏ, bất sản thể chai) dựa trên rối loạn di chuyển tế bào thần kinh trong thời kỳ phôi thai. Cơ chế phân tử gây ra rối loạn di chuyển này vẫn chưa được làm sáng tỏ, và nghiên cứu trong tương lai được mong đợi cùng với việc xác định gen gây bệnh.
7. Nghiên cứu mới nhất và triển vọng tương lai (Báo cáo giai đoạn nghiên cứu)
Gen gây bệnh vẫn chưa được xác định, nhưng việc phát hiện đột biến gen TREX1 2) là một manh mối mới cho thấy khả năng liên quan đến các đột biến không liên kết với nhiễm sắc thể X. Với sự phổ biến của phân tích bộ gen toàn diện (WES và WGS), việc xác định gen gây bệnh dự kiến sẽ được đẩy nhanh. Việc xác minh giả thuyết liên kết X và không liên kết X là nhiệm vụ chính trong tương lai.
Nhận biết hội chứng Aicardi biến thể (không điển hình)
Các báo cáo về biến thể không có bất sản thể chai hoàn toàn mà chỉ có mỏng (loạn sản) đang được tích lũy 2). Ngay cả trong các trường hợp không đáp ứng đủ cả ba tiêu chí, việc áp dụng khám đáy mắt chi tiết và tiêu chuẩn chẩn đoán mở rộng có thể cải thiện độ chính xác chẩn đoán.
Kang và cộng sự (2022) đã báo cáo thực hiện quang đông laser và cắt dịch kính 23G cho một trường hợp bệnh võng mạcdịch kính hai bên trong hội chứng Aicardi, góp phần duy trì sự phát triển của nhãn cầu và bảo vệ chức năng thị giác 2). Cũng được ghi nhận rằng các lacuna hắc võng mạc mới xuất hiện sau phẫu thuật, cho thấy phẫu thuật mắt cũng có thể góp phần xác nhận chẩn đoán.
Ngày càng có sự công nhận rằng sàng lọc đáy mắt sớm và can thiệp nhanh chóng rất quan trọng để bảo vệ chức năng võng mạc2). Laser vào các vùng vô mạch ngoại vi có khả năng ngăn ngừa sự tiến triển của bệnh võng mạc tăng sinh, và hy vọng sẽ tích lũy thêm nhiều trường hợp trong tương lai.
Nghiên cứu về việc sử dụng cannabidiol (CBD) và liệu pháp ăn kiêng ketogenic để kiểm soát động kinh đang được tiến hành 1). Hy vọng sẽ thiết lập các lựa chọn điều trị mới cho động kinh kháng trị.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.