Aicardi sendromu, ilk kez 1967’de Fransız nörolog Jean Aicardi tarafından tanımlanan konjenital nadir bir hastalıktır. X’e bağlı dominant kalıtım olduğu düşünülür ve neredeyse tüm hastalar kızdır. Erkeklerde hemizigot form öldürücü olduğundan, sadece XXY karyotipli (Klinefelter sendromu) birkaç erkek vaka bildirilmiştir.
İnsidans yaklaşık 1:110.000 doğum olarak tahmin edilmekte olup dünya çapında yaklaşık 4.000 hasta olduğu düşünülmektedir1). Tüm vakalar de novo mutasyondur ve ebeveynden çocuğa geçiş bildirilmemiştir; kardeşlerde tekrarlama riski %1’den azdır1).
Klasik triad aşağıdakilerden oluşur1):
İnfantil spazmlar (infantile spasms): Genellikle 3-4 aylıkken başlar.
Koryoretinal lakünler (chorioretinal lacunae): Bilateral yuvarlak fundus lezyonları. Bu hastalığa özgü bulgu.
Korpus kallozum agenezisi (agenesis of the corpus callosum): Kısmi veya tam yokluk.
Prognoz kötüdür. Ortalama yaşam süresi 18 yıldır ve 27 yaşına kadar hayatta kalma olasılığı %0.62 olarak bildirilmiştir1).
QAicardi sendromu erkek çocuklarda da görülür mü?
A
Bu hastalık neredeyse sadece kız çocuklarında görülür. X’e bağlı dominant kalıtım olduğu düşünülür ve erkeklerde hemizigot olduğu için öldürücüdür. Ancak, dünyada XXY karyotipli (Klinefelter sendromu) erkek çocuklarda birkaç vaka bildirilmiştir.
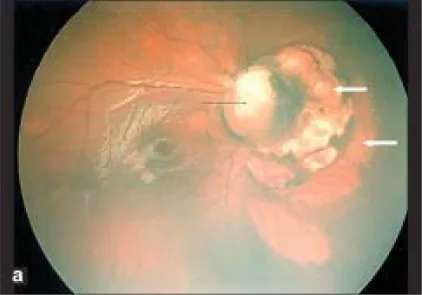
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Sağ gözün Retcam fotoğrafı, optik disk kolobomu (siyah ok) ve optik diskin nazalinde keskin sınırlı soluk alanların kubbe şeklinde odaklarını göstermektedir; bu koryoretinal lakünleri düşündürür (beyaz oklar).
Bu hastalığın ilk belirtisi genellikle 3-4 aylıkken ortaya çıkan infantil spazmlardır. Epilepsi sıklıkla tedaviye dirençli hale gelir ve çeşitli nöbet tipleri eşlik eder.
Epileptik nöbetler: İnfantil spazmlarla başlar ve tedaviye dirençli hale gelir. Bir vakada 4 aylıkken jeneralize konvülsiyonlar günde 3-4 kez (her biri 20-25 dakika süren) ortaya çıkmıştır1). Başka bir vakada 1 aylıktan itibaren sık göz kırpma nöbetleri gözlenmiştir2).
Psikomotor gelişim geriliği: Ağır zihinsel engellilik eşlik eder, bağımsız hareket ve dil edinimi genellikle zordur.
Gastrointestinal sistem disfonksiyonu: Kabızlık gibi gastrointestinal semptomlar vakaların %90’ından fazlasında mevcuttur1).
Görme bozukluğu: Fundus lezyonları, korpus kallozum agenezisi ve kortikal malformasyonlara bağlı görme azlığı oluşur.
Bu hastalığın oftalmolojik bulguları arasında, koroid-retinal lakuna (choroidal-retinal lacuna) bu hastalık için patognomonik (pathognomonic) bir bulgu olarak kabul edilir.
Koroid-Retinal Lakuna
Dağılım: Bilateral. Optik disk çevresinde ve arka kutupta yoğunlaşır, ancak perifere de uzanır.
Görünüm: Yuvarlak ila oval, sarı-beyaz ila pembe renkli lezyonlar. Prevalans %70-901).
Periferik avasküler retina: 360 derecelik avasküler bir bant oluşabilir2).
Trakşiyonel retina dekolmanı (TRD): Vasküler sap dokusu (stalk tissue) ile birlikte ortaya çıkabilir2).
Olgu sunumlarında, sağ gözde preretinal hemoraji ve 360 derece periferik avasküler bant, sol gözde ise sap dokusu (fibrovasküler sap) ve traksiyonel retina dekolmanı olan bir örnek kaydedilmiştir2). Ayrıca kortikal görme bozukluğuna (cortical visual impairment; CVI) neden olabilir3).
Korpus kallozum agenezisi: Tüm olgularda kısmi veya tam korpus kallozum yokluğu mevcuttur1). Korpus kallozum incelmesi (dysgenesis) de bir varyant olarak bildirilmiştir2).
Serebral kortikal malformasyon: Polimikrogiri (Barkovich sınıflaması Tip 2), periventriküler gri madde nodülleri ve multiloküle kistler MRG’de doğrulanır1).
EEG bulguları: Yüksek voltajlı polimorfik ritim ve multifokal diken-dalga deşarjları ile karakteristik bir patern gösterir1).
İskelet anomalileri: %40-60 oranında torakal vertebra füzyonu (T9-T10) ve kelebek vertebra (T8) gibi bulgular saptanır1).
QKoryoretinal lakünler zamanla değişir mi?
A
İlerleyici olabilir. Oküler cerrahi müdahale sonrası lakünlerin yeni görünür hale geldiği veya zamanla boyut ve sayılarının arttığı bildirilmiştir2). Düzenli fundus muayenesi ile takip önemlidir.
Aicardi sendromunun X’e bağlı dominant kalıtıldığı düşünülmektedir, ancak sorumlu gen henüz tanımlanmamıştır1). Tüm vakalar de novo mutasyonlardır ve ailesel görülme temelde yoktur. Kardeşlerde tekrarlama riski %1’den azdır ve genetik danışmanlık sonrası sonraki çocuk planlaması önerilir1).
Son yıllardaki vaka raporlarında, bir vakada TREX1 gen mutasyonu (c.292_293insA, p.(Cys99Metfs)) tespit edilmiştir2). TREX1, kromozom 3 üzerinde yer alır ve X’e bağlı hipoteziyle kısmen çeliştiğinden, sorumlu genin yeri tartışmalıdır.
Kalıtım şekli: X’e bağlı dominant (varsayılan). Hemizigot erkekler embriyonik dönemde öldürücüdür.
Mutasyonun doğası: Tümü de novo mutasyon. Kalıtsal (ebeveynden çocuğa geçiş) yoktur.
Aicardi sendromu tanısı, sorumlu gen henüz tanımlanmadığı için esas olarak klinik tanıya dayanır. Aşağıdaki klasik triadın doğrulanması tanının merkezini oluşturur1).
İnfantil spazmlar (infantile spasms)
Koryoretinal lakünalar (chorioretinal lacunae)
Korpus kallozum agenezisi (agenesis of the corpus callosum)
Üçlüden sadece ikisi mevcut olsa bile, 1999’da oluşturulan genişletilmiş tanı kriterleri ile tanı mümkündür.
Genişletilmiş tanı kriterlerinin bileşenleri aşağıda gösterilmiştir.
Multidisipliner iş birliği: Tanı için nöroloji, göz hastalıkları, ortopedi ve genetik bölümlerinin iş birliği gereklidir 1).
QGenetik test ile kesin tanı konulabilir mi?
A
Şu anda kesin tanı koyduran bir nedensel gen tanımlanmamıştır, bu nedenle yalnızca genetik test ile kesin tanı konulamaz 1). Tanı, klinik semptomlar, fundus bulguları ve görüntüleme bulgularının birleşimiyle yapılan klinik tanıya dayanır. Kapsamlı genomik analizdeki ilerlemeler, gelecekte nedensel genin tanımlanmasını umut vaat etmektedir.
Kesin bir tedavi yoktur. Tedavinin hedefleri üç ana sütundan oluşur: epilepsi kontrolü, göz komplikasyonlarının yönetimi ve rehabilitasyon yoluyla gelişimsel destek.
Epilepsi yönetimi
Birinci basamak ilaçlar: Fenitoin, levetirasetam, klobazam vb. kullanılır 1).
Dirençli vakalar: Kannabidiol (CBD), ketojenik diyet, korpus kallozotomi ve vagus sinir stimülasyonu denenir 1).
Oftalmik komplikasyonlara yönelik cerrahi müdahale raporları sunulmaktadır.
Göz
Bulgular
Tedavi
Sağ göz
Preretinal kanama ve 360 derece avasküler alan
Lazer fotokoagülasyon
Sol göz
Stalk doku ve traksiyonel retina dekolmanı (TRD)
23G vitrektomi
Vitrektomi sonrası aksiyel uzunlukta büyüme düzelmesi gözlenmiştir; sol gözde doğumdan 1 ay sonra 17.45 mm olan aksiyel uzunluk, 26. ayda 24.41 mm’ye ulaşmıştır 2). Ayrıca, ameliyat sonrası yeni koroidal lakünlerin görünür hale geldiği ve tanının kesinleşmesine katkıda bulunduğu vakalar bildirilmiştir 2).
QOküler cerrahi tedavi mümkün müdür?
A
Mümkündür. Periferik avasküler retinaya lazer fotokoagülasyon veya traksiyonel retina dekolmanına 23G vitrektomi etkili olabilir 2). Ameliyat sonrası göz büyümesinin hızlandığına dair raporlar vardır. Ancak vaka sayısı azdır ve uzman merkezlerde dikkatli yönetim gerektirir.
X kromozomunda de novo mutasyonun bu hastalığın nedeni olduğu varsayılmaktadır. X kromozomu inaktivasyonu (lionizasyon) paterni, aynı mutasyonda fenotipik çeşitliliğe yol açar. Erkeklerde hemizigot olduğu için embriyonik dönemde ölümcüldür ve yalnızca XXY karyotipli erkekler yaşayabilir.
Son yıllarda, bu hastalık tanısı konan bir vakada TREX1 gen mutasyonu (c.292_293insA, p.(Cys99Metfs)) tespit edilmiştir 2). TREX1 kromozom 3 üzerinde yer aldığından, X’e bağlı hipotezle çelişebilir ve X’e bağlı olmayan neden genlerinin varlığına işaret edebilir.
Kortikal görme bozukluğu (CVI), polimikrogiri, heterotopik gri cevher ve korpus kallozum agenezisi gibi beyin yapısal anomalilerine bağlı edinsel görme fonksiyon azalması olarak ortaya çıkar 3).
Beyin malformasyonları (polimikrogiri, korpus kallozum agenezisi) embriyonik dönemde nöronal migrasyon bozukluğuna dayanır. Bu migrasyon bozukluğunun altında yatan moleküler mekanizma henüz aydınlatılamamıştır ve neden geninin tanımlanmasıyla birlikte gelecekteki araştırmalar beklenmektedir.
7. En yeni araştırmalar ve geleceğe bakış (Araştırma aşamasındaki raporlar)
Neden olan gen henüz tanımlanmamıştır, ancak TREX1 gen mutasyonunun saptanması 2), X’e bağlı olmayan mutasyonların rol oynayabileceğini gösteren yeni bir ipucudur. Kapsamlı genom analizinin (WES ve WGS) yaygınlaşmasıyla, neden olan genin tanımlanmasının hızlanması beklenmektedir. X’e bağlı ve X’e bağlı olmayan hipotezlerin doğrulanması gelecekteki ana zorluklardan biridir.
Korpus kallozumun tamamen yok olmadığı, sadece incelme (dizgenez) gösteren mutant varyantlara ilişkin raporlar birikmektedir 2). Üçlü bulguların tümünü karşılamayan vakalarda bile ayrıntılı fundus muayenesi ve genişletilmiş tanı kriterlerinin uygulanması ile tanı doğruluğu artabilir.
Kang ve ark. (2022), Aicardi sendromlu bir bilateral vitreoretinopati vakasında lazer fotokoagülasyon ve 23G vitrektomi uyguladıklarını ve bunun göz küresi büyümesinin korunmasına ve görsel fonksiyonun korunmasına katkıda bulunduğunu bildirmiştir 2). Ayrıca ameliyat sonrası yeni koroid-retinal lakunaların görünür hale geldiği kaydedilmiş olup, oftalmik cerrahinin kesin tanıya da katkıda bulunabileceğini göstermiştir.
Erken fundus taraması ve hızlı müdahalenin retina fonksiyonunun korunması için önemli olduğu yönündeki farkındalık artmaktadır 2). Periferik avasküler alanlara lazer uygulamasının proliferatif retinopatinin ilerlemesini önleme potansiyeli vardır ve gelecekte daha fazla vaka birikimi umulmaktadır.
Kannabidiol (CBD) ve ketojenik diyetin epilepsi kontrolünde kullanımına yönelik araştırmalar ilerlemektedir 1). Dirençli epilepsi için yeni tedavi seçeneklerinin oluşturulması beklenmektedir.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.