脉络膜视网膜陷窝
分布:双侧性。密集于视乳头周围和后极部,但也延伸至周边部。
外观:圆形至椭圆形,黄白色至粉红色病变。患病率为70–90%1)。
组织学:视网膜色素上皮(RPE)缺损,从脉络膜层延伸至裸露的巩膜层2)。
病程:术后可能随时间推移大小和数量增加2)。

艾卡迪综合征是一种先天性罕见疾病,1967年由法国神经科医生Jean Aicardi首次描述。推测为X连锁显性遗传,几乎所有患者均为女婴。男性为半合子致死,因此仅在XXY核型(克氏综合征)的男性中有少数病例报道。
估计发病率约为1:110,000出生,全球患病人数约为4,000人1)。所有病例均为新发突变,无亲子传播,兄弟姐妹复发风险低于1%1)。
经典三联征包括以下三项1):
预后不良。平均生存年龄为18岁,存活至27岁的概率据报道为0.62% 1)。
本病几乎只发生在女婴中。推测为X连锁显性遗传,因为男性半合子致死。然而,全球有少数几例报告发生在具有XXY核型(克氏综合征)的男孩中。
本病的首发症状通常是在出生后3-4个月左右出现的婴儿痉挛症。癫痫常发展为药物难治性,并伴有多种发作类型。
在本病的眼科检查所见中,脉络膜视网膜陷窝被认为是具有病理特异性的(pathognomonic)表现。
脉络膜视网膜陷窝
分布:双侧性。密集于视乳头周围和后极部,但也延伸至周边部。
外观:圆形至椭圆形,黄白色至粉红色病变。患病率为70–90%1)。
组织学:视网膜色素上皮(RPE)缺损,从脉络膜层延伸至裸露的巩膜层2)。
病程:术后可能随时间推移大小和数量增加2)。
其他眼部表现
病例报告中记录了一例右眼视网膜前出血和360度周边无血管区,左眼有stalk tissue(纤维血管茎)和牵引性视网膜脱离2)。此外,也可能导致皮质视觉障碍(cortical visual impairment; CVI)3)。
可能会进展。有报道称眼科手术干预后出现新的陷窝,或陷窝的大小和数量随时间增加2)。定期眼底检查随访很重要。
Aicardi综合征被认为是X连锁显性遗传,但致病基因尚未确定1)。所有病例均为新生突变,基本无家族内发病。兄弟姐妹的复发风险低于1%,建议在遗传咨询后考虑生育计划1)。
近年的一例病例报告在一名患者中检测到TREX1基因突变(c.292_293insA, p.(Cys99Metfs))2)。TREX1位于3号染色体,与X连锁假说部分矛盾,因此致病基因的定位仍有争议。
由于致病基因尚未确定,Aicardi综合征的诊断主要依靠临床诊断。确认以下经典三联征是诊断的核心1)。
即使仅满足三联征中的两项,也可根据1999年制定的扩展诊断标准进行诊断。
扩展诊断标准的构成如下所示。
| 分类 | 主要项目 |
|---|---|
| 主要特征 | 视神经缺损、皮质畸形、异位灰质、颅内囊肿、脉络丛乳头状瘤 |
| 支持特征 | 椎肋骨异常、小眼球症、分离脑电图、半球不对称、血管畸形 |
三联征中的两项 + 两项或以上主要特征或支持特征即可确诊。
目前尚未确定可确诊的致病基因,因此仅凭基因检测无法确诊1)。主要依靠综合临床症状、眼底表现和影像学表现的临床诊断。随着全面基因组分析的进步,未来有望确定致病基因。
尚无根治性治疗方法。治疗目标包括三大支柱:癫痫控制、眼科并发症处理以及通过康复训练促进发育。
癫痫管理
一线药物:使用苯妥英、左乙拉西坦、氯巴占等1)。
难治性病例:尝试大麻二酚(CBD)、生酮饮食疗法、胼胝体切开术、迷走神经刺激术1)。
眼科干预
激光光凝:对周边无血管视网膜进行激光光凝,以预防增殖性视网膜病变的进展2)。
玻璃体切除术:对牵拉性视网膜脱离(TRD)行23G玻璃体切除术2)。
康复
早期开始:诊断后立即开始物理治疗、作业治疗、言语治疗和视觉治疗1)。
低视力护理:提供视觉障碍的辅助器具和环境调整。
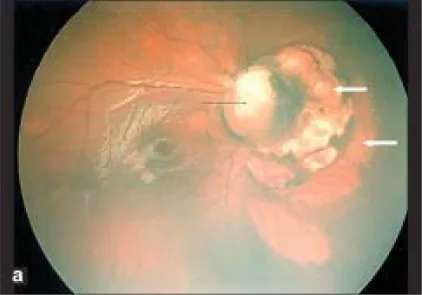
介绍眼科并发症手术干预的病例报告。
| 眼 | 所见 | 治疗 |
|---|---|---|
| 右眼 | 视网膜前出血、无血管区360度 | 激光光凝 |
| 左眼 | stalk组织、TRD | 23G玻璃体切除术 |
玻璃体切除术后确认眼轴长度恢复生长;左眼在出生后1个月时眼轴长度为17.45毫米,到26个月时增长至24.41毫米2)。此外,有报告称术后脉络膜视网膜陷窝新显现,有助于确诊2)。
可能。对周边无血管视网膜进行激光光凝,或对牵拉性视网膜脱离进行23G玻璃体切除术可能有效2)。有报告称术后眼球生长加速。但病例数较少,需要在专业机构谨慎处理。
X染色体上的新生突变被认为是本病的病因。X染色体失活(莱昂化)的模式可能导致相同突变产生表型多样性。男性为半合子状态,因此在胚胎期致死,只有XXY核型的男性才能存活。
近年来,在一例确诊为本病的患者中检测到TREX1基因突变(c.292_293insA,p.(Cys99Metfs))2)。由于TREX1位于3号染色体上,这可能与X连锁假说矛盾,提示可能存在非X连锁的致病基因。
脉络膜视网膜腔隙的组织学特征为视网膜色素上皮(RPE)缺损,从脉络膜毛细血管板延伸至裸露的巩膜层2)。缺损区域缺乏RPE和脉络膜毛细血管板,可见未分化的神经视网膜发育不良。
胎儿血管系统的持续存在被认为与纤维血管蒂组织和周边无血管视网膜的形成有关2)。这些异常血管导致牵拉性视网膜脱离。
皮质视觉障碍(CVI)是由于多小脑回、灰质异位和胼胝体缺如等脑结构异常引起的后天性视觉功能下降3)。
脑畸形(多小脑回、胼胝体缺如)基于胚胎期神经元迁移障碍。这种迁移障碍的分子机制仍不清楚,有待于致病基因的鉴定和未来的研究。
致病基因尚未明确,但TREX1基因突变的检测2)是一个新线索,提示可能涉及非X连锁突变。随着全外显子组测序(WES)和全基因组测序(WGS)的普及,有望加速致病基因的鉴定。验证X连锁假说和非X连锁假说是未来的主要课题。
关于胼胝体并非完全缺如而仅表现为变薄(发育不全)的变异型病例的报告正在积累2)。即使不完全满足三大特征的病例,通过详细的眼底检查和扩展诊断标准的应用,也可能提高诊断准确性。
Kang等人(2022)报道,对Aicardi综合征的双侧玻璃体视网膜病变进行激光光凝和23G玻璃体切除术,有助于维持眼球生长和保护视觉功能2)。他们还记录到术后脉络膜视网膜陷窝新显现,表明眼科手术也有助于确诊。
人们越来越认识到早期眼底筛查和及时干预对保护视网膜功能的重要性2)。对周边无血管区进行激光治疗可能预防增殖性视网膜病变的进展,期待未来病例的积累。
关于大麻二酚(CBD)和生酮饮食疗法用于癫痫控制的研究正在推进1)。有望为难治性癫痫建立新的治疗选择。