脈絡膜視網膜陷窩
分佈:雙側性。密集於視乳頭周圍和後極部,但也延伸至周邊部。
外觀:圓形至橢圓形,黃白色至粉紅色病變。患病率為70–90%1)。
組織學:視網膜色素上皮(RPE)缺損,從脈絡膜層延伸至裸露的鞏膜層2)。
病程:術後可能隨時間推移大小和數量增加2)。

艾卡迪症候群是一種先天性罕見疾病,1967年由法國神經科醫師Jean Aicardi首次描述。推測為X染色體顯性遺傳,幾乎所有患者均為女嬰。男性為半合子致死,因此僅在XXY核型(克氏症候群)的男性中有少數病例報告。
估計發生率約為1:110,000出生,全球罹患人數約為4,000人1)。所有病例均為新發突變,無親子傳播,兄弟姐妹復發風險低於1%1)。
經典三聯徵包括以下三項1):
預後不良。平均存活年齡為18歲,存活至27歲的機率據報導為0.62% 1)。
本病幾乎只發生在女嬰中。推測為X連鎖顯性遺傳,因為男性半合子致死。然而,全球有少數幾例報告發生在具有XXY核型(克氏症候群)的男孩中。
本病的首發症狀通常是在出生後3-4個月左右出現的嬰兒點頭痙攣。癲癇常發展為藥物難治性,並伴有多種發作類型。
在本病的眼科檢查所見中,脈絡膜視網膜陷窩被認為是具有病理特異性(pathognomonic)的表現。
脈絡膜視網膜陷窩
分佈:雙側性。密集於視乳頭周圍和後極部,但也延伸至周邊部。
外觀:圓形至橢圓形,黃白色至粉紅色病變。患病率為70–90%1)。
組織學:視網膜色素上皮(RPE)缺損,從脈絡膜層延伸至裸露的鞏膜層2)。
病程:術後可能隨時間推移大小和數量增加2)。
其他眼部表現
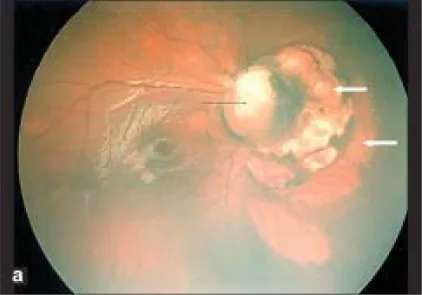
病例報告中記錄了一例右眼視網膜前出血和360度周邊無血管區,左眼有stalk tissue(纖維血管莖)和牽引性視網膜剝離2)。此外,也可能導致皮質視覺障礙(cortical visual impairment; CVI)3)。
可能會進展。有報導稱眼科手術干預後出現新的陷窩,或陷窩的大小和數量隨時間增加2)。定期眼底檢查追蹤很重要。
Aicardi症候群被認為是X染色體顯性遺傳,但致病基因尚未確定1)。所有病例均為新生突變,基本上無家族內發病。兄弟姐妹的復發風險低於1%,建議在遺傳諮詢後考慮生育計畫1)。
近年的一例病例報告在一名患者中檢測到TREX1基因突變(c.292_293insA, p.(Cys99Metfs))2)。TREX1位於3號染色體,與X染色體顯性假說部分矛盾,因此致病基因的位置仍有爭議。
由於致病基因尚未確定,Aicardi症候群的診斷主要依靠臨床診斷。確認以下經典三聯徵是診斷的核心1)。
即使僅符合三徵中的兩項,仍可根據1999年制定的擴展診斷標準進行診斷。
擴展診斷標準的組成如下所示。
| 分類 | 主要項目 |
|---|---|
| 主要特徵 | 視神經缺損、皮質畸形、異位灰質、顱內囊腫、脈絡叢乳頭狀瘤 |
| 支持特徵 | 椎肋骨異常、小眼球症、分離腦電圖、半球不對稱、血管畸形 |
三徵中的兩項 + 兩項或以上主要特徵或支持特徵即可確診。
目前尚未確定可確診的致病基因,因此僅靠基因檢測無法確診1)。主要依據臨床症狀、眼底所見和影像學所見進行臨床診斷。隨著全面性基因組分析的進步,未來有望找出致病基因。
目前無根治性治療。治療目標包括三大支柱:癲癇控制、眼科併發症處理,以及透過復健促進發展。
癲癇管理
第一線藥物:使用苯妥英、左乙拉西坦、氯巴占等1)。
難治性案例:嘗試大麻二酚(CBD)、生酮飲食療法、胼胝體切開術、迷走神經刺激術1)。
眼科介入
雷射光凝固:對周邊無血管視網膜進行雷射光凝固,以預防增殖性視網膜病變的進展2)。
玻璃體切除術:對牽引性視網膜剝離(TRD)進行23G玻璃體切除術2)。
復健
早期開始:診斷後立即開始物理治療、職能治療、語言治療和視覺治療1)。
低視力照護:提供視覺障礙的輔助器具和環境調整。
介紹眼科併發症手術介入的病例報告。
| 眼 | 所見 | 治療 |
|---|---|---|
| 右眼 | 視網膜前出血、無血管區360度 | 雷射光凝固 |
| 左眼 | stalk組織、TRD | 23G玻璃體切除術 |
玻璃體切除術後確認眼軸長度恢復生長;左眼在出生後1個月時眼軸長度為17.45毫米,到26個月時增長至24.41毫米2)。此外,有報告指出術後脈絡膜視網膜陷窩新顯現,有助於確診2)。
可能。對周邊無血管視網膜進行雷射光凝,或對牽引性視網膜剝離進行23G玻璃體切除術可能有效2)。有報告稱術後眼球生長加速。但病例數較少,需要在專業機構謹慎處理。
X染色體上的新生突變被認為是本病的病因。X染色體不活化(萊昂化)的模式可能導致相同突變產生表型多樣性。男性為半合子狀態,因此在胚胎期致死,只有XXY核型的男性才能存活。
近年來,在一例確診為本病的患者中檢測到TREX1基因突變(c.292_293insA,p.(Cys99Metfs))2)。由於TREX1位於3號染色體上,這可能與X連鎖假說矛盾,提示可能存在非X連鎖的致病基因。
脈絡膜視網膜腔隙的組織學特徵為視網膜色素上皮(RPE)缺損,從脈絡膜微血管板延伸至裸露的鞏膜層2)。缺損區域缺乏RPE和脈絡膜微血管板,可見未分化的神經視網膜發育不良。
胎兒血管系統的持續存在被認為與纖維血管蒂組織和周邊無血管視網膜的形成有關2)。這些異常血管導致牽引性視網膜剝離。
皮質視覺障礙(CVI)是由於多小腦迴、灰質異位和胼胝體缺如等腦結構異常引起的後天性視覺功能下降3)。
腦畸形(多小腦迴、胼胝體缺如)基於胚胎期神經元遷移障礙。這種遷移障礙的分子機制仍不清楚,有待於致病基因的鑑定和未來的研究。
致病基因尚未明確,但TREX1基因突變的檢測2)是一個新線索,提示可能涉及非X連鎖突變。隨著全外顯子組測序(WES)和全基因組測序(WGS)的普及,有望加速致病基因的鑑定。驗證X連鎖假說和非X連鎖假說是未來的主要課題。
關於胼胝體並非完全缺如而僅表現為變薄(發育不全)的變異型病例的報告正在累積2)。即使不完全滿足三大特徵的病例,透過詳細的眼底檢查和擴展診斷標準的應用,也可能提高診斷準確性。
Kang等人(2022)報告,對Aicardi症候群的雙側玻璃體視網膜病變進行雷射光凝和23G玻璃體切除術,有助於維持眼球生長和保護視覺功能2)。他們也記錄到術後脈絡膜視網膜陷窩新顯現,表明眼科手術也有助於確診。
人們越來越認識到早期眼底篩查和及時介入對保護視網膜功能的重要性2)。對周邊無血管區進行雷射治療可能預防增殖性視網膜病變的進展,期待未來病例的累積。
關於大麻二酚(CBD)和生酮飲食療法用於癲癇控制的研究正在推進1)。有望為難治性癲癇建立新的治療選擇。