
視神經盤缺損
一目瞭然的要點
Section titled “一目瞭然的要點”1. 什麼是視神經盤缺損?
Section titled “1. 什麼是視神經盤缺損?”視神經盤缺損是一種先天性異常,表現為視神經盤異常擴大和邊界清晰的白色凹陷。它由眼杯裂(胚胎裂)閉合不全引起,該裂通常在胚胎第7週閉合。視網膜血管並非從一個點起始,而是從凹陷邊緣或凹陷內的不同部位起始。
當眼杯裂閉合不全局限於後部(視神經側)時,發生視神經盤缺損。如果閉合不全從前到後更廣泛,則形成虹膜至脈絡膜缺損的譜系。在整個缺損譜系中,視神經盤缺損對應於眼杯裂閉合不全的後端,是虹膜缺損(前端)連續譜系的一部分,但也存在局限於視神經盤的孤立型。
ICD-10編碼為H47.319(視神經)。
與牽牛花症候群的鑑別很重要。牽牛花症候群在視盤中心可見膠質增生組織,血管呈放射狀走向。視神經盤缺損可透過無膠質增生、凹陷以下方為主以及血管從凹陷邊緣或凹陷內不同部位起始來鑑別。
2. 主要症狀與臨床所見
Section titled “2. 主要症狀與臨床所見”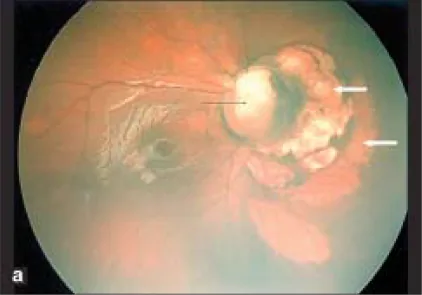
視力取決於乳頭黃斑束是否被捲入缺損及其程度。從超過1.0到不良不等,但即使黃斑部無異常,由於視神經異常,視力下降的病例也很多。
- 視力下降:取決於乳頭黃斑束的捲入程度。視力不良病例可能出現廢用性斜視。
- 視野缺損:常對應於乳頭下方的缺損,產生上方視野缺損。
- 斜視:視力不良病例可繼發廢用性斜視。
眼底可見以視神經乳頭下方為中心的缺損,以及眼球下方的視網膜脈絡膜缺損。伴有血管走行異常,由於視網膜中央動脈在乳頭後方分支,可見多條視網膜動脈從乳頭發出。乳頭上方的邊緣常殘留,即使整體凹陷,典型情況下下方比上方更明顯。
乳頭區域凹陷,乳頭缺失或部分缺損,周圍的脈絡膜、視網膜色素上皮(RPE)和鞏膜也變薄。乳頭區域凹陷的下方,存在由於胚胎裂閉合不全導致的視網膜脈絡膜萎縮和紋理樣改變。
視神經乳頭缺損根據合併範圍分類如下。
- 單純視神經乳頭缺損型:由於眼杯裂後部侷限性閉合不全所致。
- 合併視網膜脈絡膜缺損型:顯示更廣泛範圍的眼杯裂閉合不全。
- 虹膜/睫狀體缺損聯合型:從前端到後端連續的廣泛型。
- Fuchs缺損:輕微型,在視盤下方顯示類似圓錐的萎縮病變。視力通常相對保留。
- 常合併虹膜缺損和脈絡膜缺損。
- 當合併脈絡膜缺損且凹陷區域較大時,可能出現小眼球。
- 漿液性視網膜剝離:即使單獨視盤缺損也可能發生。
- 裂孔源性視網膜剝離:在複雜的視網膜脈絡膜缺損病例中可能繼發。
- 經鞏膜濾過導致的低眼壓:已有鞏膜缺損處房水滲漏的病例報導7)。
3. 病因、流行病學、風險因素
Section titled “3. 病因、流行病學、風險因素”患病率報導為3~8/100,000。單眼和雙眼病例比例相當,多為散發病例,但常有家族史。已報導多種遺傳方式,包括體染色體顯性、體染色體隱性和X連鎖遺傳4)。
缺損的整體遺傳診斷率仍低於30%5)。許多未發現基因突變的散發病例提示環境因素和多個修飾基因的參與。
胚胎裂(眼杯裂)在胚胎第4週神經外胚層形成眼杯時出現於腹側。第5週完成,第6週開始閉合。閉合從赤道部附近向前方(虹膜側)和後方(視神經側)進行,第7週完成。後方局部閉合不全導致視盤缺損。
| 基因 | 相關疾病 | 備註 |
|---|---|---|
| PAX2 | 腎缺損症候群(renal coloboma syndrome) | 參與眼球腹側決定和胚胎裂閉合1) |
| CHD7 | CHARGE症候群 | 第8號染色體(8q12.2),指定難治疾病 |
| FZD5 | 症候性缺損+小角膜 | Wnt訊息傳導路徑受體2) |
視神經盤缺損可能合併以下全身症候群。
- CHARGE症候群:以缺損(C)、心臟畸形(H)、後鼻孔閉鎖(A)、生長遲緩(R)、生殖器發育不全(G)、耳部異常(E)的首字母命名的多器官畸形症候群。致病基因為CHD7,被認定為指定難病。
- Aicardi症候群:伴有胼胝體缺損、癲癇、精神發育遲滯。多見於女童。缺損常表現為脈絡膜視網膜上的多個空白區(lacunae)。
- 腎缺損症候群:由PAX2基因突變引起。合併腎尿路發育異常。需要長期追蹤腎功能。PAX2的c.76delG移碼突變已在局部節段性腎小球硬化症(FSGS)家系中被鑑定,表現型譜比以往認為的更廣1)。
4. 診斷與檢查方法
Section titled “4. 診斷與檢查方法”僅憑檢眼鏡所見即可診斷。關鍵診斷要點為視盤下方為主的邊界清晰的白色凹陷及特徵性血管走向異常(從凹陷邊緣或凹陷內發出大量血管)。確診需使用超音波、MRI、CT、光學同調斷層掃描(OCT)。
雖然散發病例較多,但也可能存在家族史,因此需要仔細詢問家族史。
需進行頭部MRI/CT以檢查是否合併顱內畸形(如胼胝體缺損)。應請兒科會診以確認是否存在CHARGE症候群、Aicardi症候群等全身併發症。
| 檢查 | 目的 |
|---|---|
| 眼底檢查(散瞳) | 評估視神經盤形態、血管走向及視網膜剝離 |
| OCT(光學同調斷層掃描) | 詳細評估視神經盤及黃斑結構 |
| 超音波檢查(B模式) | 眼底觀察不佳時偵測視網膜剝離 |
| 視野檢查 | 評估視野缺損型態(如上視野缺損) |
| 頭部MRI | 檢查中樞神經系統併發症,如胼胝體缺損及腦膨出 |
| 腎臟超音波 | 篩檢腎臟缺損症候群 |
| 基因檢測 | 檢測PAX2、CHD7等基因突變(雙側或症候群性病例) |
| 鑑別診斷 | 鑑別要點 |
|---|---|
| 晨光症候群(morning glory syndrome) | 視乳頭中心有神經膠質增生組織,血管呈放射狀走向。而缺損(coloboma)為下方優勢凹陷,無神經膠質增生。 |
| 視乳頭周圍葡萄腫(peripapillary staphyloma) | 圍繞視乳頭的鞏膜向後膨隆。缺損(coloboma)是視乳頭本身的缺損。 |
| 視乳頭PFV/PHPV(原始玻璃體動脈殘留) | 伴有玻璃體索條和視網膜皺褶。眼底表現與缺損(coloboma)不同。 |
| 巨大視乳頭(megalopapilla) | 視乳頭直徑大,但形態接近正常。無凹陷或血管走向異常。5) |
| 視神經發育不全 | 視乳頭小(DM/DD比≥3.2)。缺損(coloboma)為視乳頭擴大和凹陷。 |
| 青光眼性視神經萎縮 | 進行性凹陷擴大和眼壓升高。缺損(coloboma)為非進行性,眼壓正常。 |
5. 標準治療方法
Section titled “5. 標準治療方法”視神經盤缺損本身是一種先天性結構異常,無根治性治療。主要根據併發症的有無和類型進行症狀治療。
由於是非進行性先天異常,如果沒有漿液性視網膜剝離等併發症,則繼續定期進行眼底檢查。兒童期建議每半年至一年進行一次散瞳眼底檢查。
漿液性視網膜剝離的處理
Section titled “漿液性視網膜剝離的處理”漿液性視網膜剝離的治療尚無定論,有自然消退的病例報告。有時會觀察數月。如果觀察後無改善,則考慮手術介入。
推測是由於凹陷處的結構異常導致玻璃體液進入視網膜下腔,也有研究表明凹陷與蜘蛛膜下腔相通可能導致腦脊髓液流入。
裂孔性視網膜剝離的處理
Section titled “裂孔性視網膜剝離的處理”對於裂孔性視網膜剝離,進行玻璃體手術和凹陷周圍的光凝固治療。術後視力預後不一定良好。
有報告使用纖維蛋白膠進行視網膜復位術治療缺損相關視網膜剝離,在缺損邊緣的視網膜裂孔周圍塗抹纖維蛋白膠以加強黏附 3)。部分病例最終視力改善至20/50。
對於視力不良的病例,尤其是單眼性,進行屈光矯正和遮蓋療法(遮蓋健眼)。兒童期早期介入很重要。但由於視神經本身結構異常導致的視力下降,弱視治療效果有限。
漿液性視網膜剝離有時可自然消退,因此首先進行數月的觀察。對於裂孔源性視網膜剝離,進行玻璃體手術和凹陷周圍的光凝。近年來也有使用纖維蛋白膠加強黏附的報告。但術後視力預後不一定良好。
6. 病理生理學與詳細發病機制
Section titled “6. 病理生理學與詳細發病機制”胚胎裂的閉合過程
Section titled “胚胎裂的閉合過程”眼杯在胚胎第4週由神經外胚層形成。眼杯腹側出現胚胎裂(眼杯裂),玻璃體動脈通過其中。此裂在第5週完成,第6週開始閉合。閉合從赤道部附近開始,向前方(虹膜側)和後方(視神經側)推進,第7週完成。後方局部閉合不全導致視神經缺損。
閉合過程涉及上皮-間質轉化(EMT)。胚胎裂邊緣的神經視網膜上皮細胞降解基底膜,獲得間質特性並融合。此過程障礙導致缺損6)。
PAX2基因參與眼球腹側的決定,並被認為參與胚胎裂的閉合。PAX2突變導致腎缺損症候群。在FSGS家系中發現了PAX2 c.76delG移碼突變,腎缺損症候群的表現型比以前認為的更廣泛1)。
FZD5基因編碼Wnt信號通路的受體。功能喪失型突變損害Wnt信號的配體依賴性激活,導致胚胎裂閉合不全和小角膜。呈隱性遺傳模式2)。
CHD7基因編碼染色質重塑因子,參與神經嵴細胞的分化和遷移。突變導致CHARGE症候群。
漿液性視網膜剝離的機制
Section titled “漿液性視網膜剝離的機制”推測由於凹陷部位的結構異常,玻璃體液流入視網膜下腔。也有提示透過凹陷與蜘蛛膜下腔的交通導致腦脊液流入的可能性,這被認為是漿液性視網膜剝離治療困難的原因之一。
視神經盤缺損是一種非進行性的固定先天異常。合併視網膜剝離是惡化視力預後的主要因素。即使對裂孔源性視網膜剝離進行玻璃體手術後,視力預後也往往不佳。
7. 最新研究與未來展望
Section titled “7. 最新研究與未來展望”PAX2突變表現型譜的擴展
Section titled “PAX2突變表現型譜的擴展”Hu等人(2024)報告在一個局部節段性腎小球硬化症(FSGS)家系中鑑定出PAX2的c.76delG框移突變1)。腎缺損症候群的表現型比以前認為的更廣泛,視神經盤缺損病例中腎功能篩檢的重要性再次得到證實。提示應擴大缺損患者腎功能評估的適應症。
FZD5與Wnt訊息傳導異常
Section titled “FZD5與Wnt訊息傳導異常”Cortes-Gonzalez等人(2024)報告FZD5的純合錯義突變(p.M160V)導致症候群性眼缺損和小角膜2)。功能分析證實,隱性遺傳模式下Wnt訊息的配體依賴性活化受損。缺損的遺傳學診斷率低於30%,新致病基因的鑑定有望提高診斷水準。
視網膜剝離的新手術技術
Section titled “視網膜剝離的新手術技術”Jain等人(2024)報告對缺損相關視網膜剝離施行纖維蛋白膠合併視網膜復位術,最終視力改善至20/503)。在缺損邊緣的視網膜裂孔周圍塗抹纖維蛋白膠以增強黏附的技術,作為傳統玻璃體手術加光凝的輔助選擇受到關注。病例數的累積和長期結果的驗證是未來的課題。
8. 參考文獻
Section titled “8. 參考文獻”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.