
Koloboma diskus optikus
Poin-poin penting sekilas
Section titled “Poin-poin penting sekilas”1. Apa itu Koloboma Diskus Optikus?
Section titled “1. Apa itu Koloboma Diskus Optikus?”Koloboma diskus optikus adalah kelainan kongenital yang ditandai dengan pembesaran abnormal diskus optikus dan cekungan putih dengan batas tegas. Terjadi akibat gagal menutupnya celah optik (celah embrionik) yang seharusnya menutup pada minggu ke-7 kehamilan. Pembuluh darah retina tidak berasal dari satu titik, melainkan dari berbagai lokasi di tepi atau di dalam cekungan.
Jika kegagalan penutupan celah optik terbatas pada bagian posterior (sisi saraf optik), terjadilah koloboma saraf optik. Jika terdapat kegagalan penutupan yang lebih luas dari anterior ke posterior, akan terbentuk spektrum koloboma iris–koroid. Dalam spektrum koloboma secara keseluruhan, koloboma diskus optikus merupakan ujung posterior dari kegagalan penutupan celah optik dan merupakan bagian dari spektrum kontinu yang dimulai dari koloboma iris (ujung anterior), namun terdapat pula bentuk terbatas yang hanya melibatkan diskus optikus.
Kode ICD-10 adalah H47.319 (saraf optik).
Penting untuk membedakannya dari sindrom morning glory. Pada sindrom morning glory, terdapat jaringan glial proliferatif di pusat diskus optikus, dan pembuluh darah berjalan secara radial. Pada koloboma diskus optikus, tidak ada proliferasi glial, cekungan lebih dominan di bagian bawah, dan pembuluh darah berasal dari berbagai lokasi di tepi atau di dalam cekungan, yang membedakannya.
Koloboma diskus optikus menunjukkan cekungan putih dengan batas tegas yang dominan di bagian bawah, dan pembuluh darah berasal dari berbagai lokasi di tepi atau di dalam cekungan. Tidak ada proliferasi glial. Pada sindrom morning glory, terdapat jaringan glial proliferatif di pusat diskus optikus, dan pembuluh darah berjalan secara radial dari tepi diskus. Keduanya adalah kelainan kongenital diskus optikus, namun dapat dibedakan melalui temuan fundus.
2. Gejala utama dan temuan klinis
Section titled “2. Gejala utama dan temuan klinis”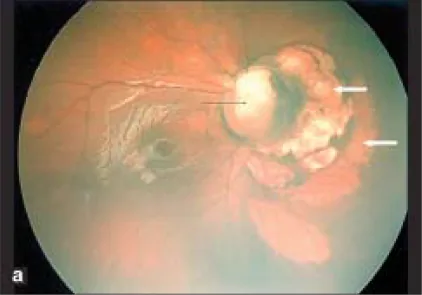
Gejala subjektif
Section titled “Gejala subjektif”Ketajaman penglihatan ditentukan oleh apakah berkas papilomakular terlibat dalam koloboma dan sejauh mana. Ketajaman bervariasi dari lebih dari 1,0 hingga kasus buruk, namun seringkali ketajaman menurun meskipun makula tidak terganggu karena kelainan saraf optik.
- Penurunan ketajaman penglihatan: Tergantung pada derajat keterlibatan berkas papilomakular. Pada kasus penglihatan buruk, dapat terjadi strabismus ambliopia.
- Defek lapang pandang: Sering terjadi defek lapang pandang superior yang sesuai dengan koloboma di bagian inferior diskus.
- Strabismus: Pada kasus penglihatan buruk, strabismus ambliopia dapat terjadi sekunder.
Temuan fundus
Section titled “Temuan fundus”Pada fundus, terjadi defek pada diskus optikus dan koroid-retina di bagian inferior bola mata. Disertai kelainan jalur pembuluh darah, dan karena arteri retina sentral bercabang di belakang diskus, banyak arteri retina tampak keluar dari diskus. Batas superior diskus sering tersisa, dan meskipun seluruh diskus cekung, secara khas bagian inferior lebih dalam dibandingkan superior.
Area diskus cekung, diskus tidak ada atau sebagian hilang, dan koroid sekitarnya, epitel pigmen retina, serta sklera tipis. Di bawah cekungan area diskus, terdapat atrofi koroid-retina dan tampilan bergaris akibat gagal menutupnya fisura embrional.
Klasifikasi
Section titled “Klasifikasi”Koloboma diskus optikus diklasifikasikan berdasarkan luasnya keterlibatan sebagai berikut.
- Tipe koloboma diskus optikus saja: Disebabkan oleh gagal menutupnya fisura optik bagian posterior secara lokal.
- Tipe dengan koloboma koroid-retina: Menunjukkan gagal menutupnya fisura optik yang lebih luas.
- Tipe gabungan koloboma iris-badan siliaris: Tipe luas yang berkesinambungan dari ujung depan ke belakang.
- Koloboma Fuchs: Tipe ringan, menunjukkan lesi atrofi mirip konus di bawah papil. Penglihatan sering relatif terjaga.
Komplikasi
Section titled “Komplikasi”Komplikasi okular
Section titled “Komplikasi okular”- Sering disertai koloboma iris dan koloboma koroid.
- Jika disertai koloboma koroid dan area cekungan luas, dapat menunjukkan mikroftalmus.
- Ablasio retina serosa: Dapat terjadi bahkan pada koloboma diskus optikus saja.
- Ablasio retina regmatogen: Dapat terjadi sekunder pada kasus koloboma retina-koroid yang kompleks.
- Hipotoni akibat filtrasi transsklera: Telah dilaporkan kasus kebocoran humor akuus melalui defek sklera 7).
3. Penyebab, Epidemiologi, Faktor Risiko
Section titled “3. Penyebab, Epidemiologi, Faktor Risiko”Epidemiologi
Section titled “Epidemiologi”Prevalensi dilaporkan 3–8/100.000. Unilateral dan bilateral dianggap sama, sebagian besar kasus sporadis, tetapi sering ada riwayat keluarga. Telah dilaporkan berbagai pola pewarisan seperti autosomal dominan, autosomal resesif, dan terkait-X 4).
Tingkat diagnosis genetik untuk seluruh koloboma kurang dari 30% 5). Banyak kasus sporadis tanpa mutasi gen yang teridentifikasi, dan diduga melibatkan faktor lingkungan serta beberapa gen modifier.
Mekanisme terjadinya
Section titled “Mekanisme terjadinya”Fisura embrionik (fisura optik) terjadi pada minggu ke-4 kehamilan saat pembentukan cawan optik dari neuroektoderm di sisi ventral. Selesai pada minggu ke-5, dan penutupan dimulai pada minggu ke-6. Penutupan berlangsung dari daerah ekuator ke anterior (arah iris) dan posterior (arah saraf optik), selesai pada minggu ke-7. Jika penutupan posterior tidak sempurna dan terbatas, terjadi koloboma saraf optik.
Gen terkait
Section titled “Gen terkait”| Gen | Penyakit terkait | Catatan |
|---|---|---|
| PAX2 | Sindrom renal coloboma | Berperan dalam penentuan ventral mata dan penutupan fisura embrionik1) |
| CHD7 | Sindrom CHARGE | Kromosom 8 (8q12.2), penyakit langka yang ditetapkan |
| FZD5 | Koloboma simptomatik + mikrokornea | Reseptor jalur sinyal Wnt2) |
Komplikasi sistemik
Section titled “Komplikasi sistemik”Koloboma diskus optikus dapat menyertai sindrom sistemik berikut.
- Sindrom CHARGE: Sindrom malformasi multi-organ yang diambil dari huruf pertama: Koloboma (C), Kelainan jantung (H), Atresia koana (A), Retardasi pertumbuhan (R), Hipoplasia genital (G), Kelainan telinga (E). Gen penyebab adalah CHD7, dan ditetapkan sebagai penyakit langka tertentu.
- Sindrom Aicardi: Ditandai dengan agenesis korpus kalosum, epilepsi, dan keterbelakangan mental. Lebih sering terjadi pada anak perempuan. Koloboma sering muncul sebagai banyak celah (lacunae) di koroid dan retina.
- Sindrom koloboma ginjal: Disebabkan oleh mutasi gen PAX2. Berhubungan dengan malformasi saluran kemih dan ginjal. Diperlukan pemantauan jangka panjang untuk gangguan fungsi ginjal. Mutasi pergeseran kerangka c.76delG pada PAX2 telah diidentifikasi pada keluarga dengan glomerulosklerosis segmental fokal (FSGS), menunjukkan spektrum fenotip yang lebih luas dari yang diperkirakan sebelumnya 1).
Koloboma diskus optikus dapat berhubungan dengan sindrom sistemik seperti Sindrom CHARGE (mutasi CHD7, malformasi multi-organ, penyakit langka tertentu), Sindrom Aicardi (agenesis korpus kalosum, epilepsi, dominasi perempuan), dan Sindrom koloboma ginjal (mutasi PAX2, malformasi saluran kemih dan ginjal). Pada kasus bilateral atau jika ada temuan sistemik, disarankan konsultasi pediatri dan konseling genetik.
4. Diagnosis dan Metode Pemeriksaan
Section titled “4. Diagnosis dan Metode Pemeriksaan”Metode Diagnosis
Section titled “Metode Diagnosis”Diagnosis dapat ditegakkan hanya dengan temuan oftalmoskopi. Cekungan putih dengan batas tegas di bagian inferior diskus dan kelainan jalur pembuluh darah yang khas (banyak pembuluh darah berasal dari tepi atau dalam cekungan) adalah poin kunci diagnosis. Untuk konfirmasi, digunakan ultrasonografi, MRI, CT, dan tomografi koherensi optik (OCT).
Meskipun sebagian besar kasus bersifat sporadis, mungkin ada riwayat keluarga, sehingga anamnesis riwayat keluarga yang cermat diperlukan.
Untuk mendeteksi malformasi intrakranial yang menyertai (seperti agenesis korpus kalosum), diperlukan MRI/CT kepala. Konsultasi pediatri dilakukan untuk memeriksa kemungkinan komplikasi sistemik seperti Sindrom CHARGE atau Sindrom Aicardi.
Pemeriksaan
Section titled “Pemeriksaan”| Pemeriksaan | Tujuan |
|---|---|
| Pemeriksaan fundus (dengan dilatasi pupil) | Evaluasi morfologi diskus optikus, jalur pembuluh darah, dan ablasi retina |
| OCT (Optical Coherence Tomography) | Evaluasi detail struktur diskus optikus dan makula |
| Ultrasonografi (B-mode echo) | Deteksi ablasi retina pada kasus dengan visualisasi fundus yang buruk |
| Tes lapang pandang | Evaluasi pola defek lapang pandang (misalnya defek lapang pandang superior) |
| MRI kepala | Deteksi komplikasi saraf pusat seperti agenesis korpus kalosum dan ensefalokel |
| USG ginjal | Skrining sindrom koloboma ginjal |
| Tes genetik | Pencarian mutasi pada PAX2, CHD7, dll. (pada kasus bilateral dan sindromik) |
Diagnosis banding
Section titled “Diagnosis banding”| Penyakit Diferensial | Poin Diferensial |
|---|---|
| Sindrom morning glory (morning glory syndrome) | Jaringan glial di pusat papil, pembuluh darah berjalan radial. Pada koloboma, cekungan dominan di bawah dan tidak ada proliferasi glial |
| Stafiloma peripapiler (peripapillary staphyloma) | Penonjolan posterior sklera yang mengelilingi papil. Koloboma adalah defek pada papil itu sendiri |
| PFV/PHPV papil (persisten arteri hialoid primordial) | Disertai tali vitreus dan lipatan retina. Berbeda dengan koloboma pada gambaran fundus |
| Megalopapilla (megalopapilla) | Diameter papil besar tetapi bentuk mendekati normal. Tidak ada cekungan atau kelainan jalur pembuluh darah5) |
| Hipoplasia saraf optik | Papil kecil (rasio DM/DD ≥ 3.2). Koloboma menyebabkan papil membesar dan cekung |
| Atrofi saraf optik glaukomatosa | Perluasan cekungan progresif dan peningkatan tekanan intraokular. Koloboma non-progresif dengan tekanan intraokular normal |
5. Metode pengobatan standar
Section titled “5. Metode pengobatan standar”Koloboma diskus optikus adalah kelainan struktural bawaan dan tidak ada pengobatan kuratif. Pengobatan simtomatik berdasarkan ada tidaknya dan jenis komplikasi menjadi fokus utama.
Observasi
Section titled “Observasi”Karena merupakan kelainan bawaan non-progresif, jika tidak ada komplikasi seperti ablasi retina serosa, pemeriksaan fundus secara teratur dilanjutkan. Pada masa kanak-kanak, pemeriksaan oftalmoskopi dengan pupil dilatasi setiap 6 bulan hingga 1 tahun dianjurkan.
Penanganan ablasi retina serosa
Section titled “Penanganan ablasi retina serosa”Tidak ada konsensus mengenai pengobatan ablasi retina serosa, dan beberapa kasus dilaporkan mengalami resolusi spontan. Observasi selama beberapa bulan dapat dilakukan. Jika tidak ada perbaikan setelah observasi, intervensi bedah dipertimbangkan.
Diduga mekanisme masuknya humor vitreus ke ruang subretina akibat kelainan struktural pada cekungan, dan kemungkinan masuknya cairan serebrospinal melalui hubungan antara cekungan dan ruang subarachnoid juga disarankan.
Penanganan ablasi retina regmatogen
Section titled “Penanganan ablasi retina regmatogen”Untuk ablasi retina regmatogen, dilakukan vitrektomi dan fotokoagulasi di sekitar cekungan. Prognosis visual pascaoperasi belum tentu baik.
Teknik reposisi retina menggunakan lem fibrin telah dilaporkan untuk ablasi retina terkait koloboma, di mana lem fibrin dioleskan di sekitar robekan retina pada tepi koloboma untuk memperkuat adhesi 3). Beberapa kasus menunjukkan perbaikan ketajaman visual akhir menjadi 20/50.
Terapi ambliopia
Section titled “Terapi ambliopia”Pada kasus dengan penglihatan buruk, terutama unilateral, dilakukan koreksi refraksi dan terapi oklusi (menutup mata sehat). Intervensi dini pada masa kanak-kanak penting. Namun, pada penurunan penglihatan akibat kelainan struktural saraf optik itu sendiri, efek terapi ambliopia terbatas.
Manajemen Sistemik
Section titled “Manajemen Sistemik”- Kasus dengan sindrom CHARGE: memerlukan manajemen multidisiplin dengan bedah jantung, THT, endokrinologi, dll.
- Sindrom renal-coloboma: lakukan pemantauan jangka panjang fungsi ginjal.
- Konseling genetik: direkomendasikan pada kasus bilateral atau sindromik.
Ablasi retina serosa dapat mengalami resolusi spontan, sehingga pertama-tama dilakukan observasi selama beberapa bulan. Untuk ablasi retina regmatogen, dilakukan vitrektomi dan fotokoagulasi di sekitar area cekung. Dalam beberapa tahun terakhir, telah dilaporkan penguatan adhesi dengan menggunakan lem fibrin. Namun, prognosis visual pascaoperasi tidak selalu baik.
6. Fisiopatologi dan Mekanisme Terperinci
Section titled “6. Fisiopatologi dan Mekanisme Terperinci”Proses Penutupan Fisura Embrio
Section titled “Proses Penutupan Fisura Embrio”Cawan optik terbentuk dari neuroektoderm pada minggu ke-4 kehamilan. Fisura embrio (fisura cawan optik) muncul di sisi ventral cawan optik, dan arteri hialoid melewatinya. Fisura ini selesai terbentuk pada minggu ke-5, dan penutupan dimulai pada minggu ke-6. Penutupan dimulai dari dekat ekuator dan berlanjut ke anterior (sisi iris) dan posterior (sisi saraf optik), selesai pada minggu ke-7. Penutupan posterior yang tidak sempurna secara lokal menyebabkan koloboma saraf optik.
Proses penutupan melibatkan transisi epitel-mesenkim (EMT). Sel-sel epitel neurosensori retina di tepi fisura embrio mendegradasi membran basal, memperoleh fenotip mesenkimal, dan kemudian menyatu. Gangguan pada proses ini menyebabkan koloboma6).
Mekanisme Molekuler
Section titled “Mekanisme Molekuler”Gen PAX2 terlibat dalam penentuan sisi ventral mata dan diyakini berperan dalam penutupan fisura embrio. Mutasi PAX2 menyebabkan sindrom renal-coloboma. Mutasi pergeseran kerangka c.76delG pada PAX2 telah diidentifikasi pada keluarga dengan FSGS, dan fenotip sindrom renal-coloboma lebih luas dari yang diperkirakan sebelumnya1).
Gen FZD5 mengkode reseptor untuk jalur sinyal Wnt. Mutasi kehilangan fungsi menyebabkan gangguan aktivasi sinyal Wnt yang bergantung pada ligan, mengakibatkan penutupan fisura embrio yang tidak sempurna dan mikrokornea. Mengikuti pola pewarisan resesif2).
Gen CHD7 mengkode faktor remodeling kromatin dan terlibat dalam diferensiasi dan migrasi sel krista neuralis. Mutasi menyebabkan sindrom CHARGE.
Mekanisme Ablasi Retina Serosa
Section titled “Mekanisme Ablasi Retina Serosa”Diperkirakan bahwa kelainan struktural pada area cekungan menyebabkan cairan vitreus mengalir ke ruang subretina. Kemungkinan masuknya cairan serebrospinal melalui hubungan antara cekungan dan ruang subarachnoid juga telah disarankan, dan ini dianggap sebagai salah satu faktor yang membuat pengobatan ablasi retina serosa menjadi sulit.
Prognosis dan Perjalanan Penyakit
Section titled “Prognosis dan Perjalanan Penyakit”Koloboma diskus optikus adalah kelainan bawaan yang tetap dan tidak progresif. Komplikasi ablasi retina merupakan faktor utama yang memperburuk prognosis visual. Bahkan setelah vitrektomi untuk ablasi retina regmatogen, prognosis visual seringkali tidak dapat diharapkan baik.
7. Penelitian Terbaru dan Prospek Masa Depan
Section titled “7. Penelitian Terbaru dan Prospek Masa Depan”Perluasan Spektrum Fenotip Mutasi PAX2
Section titled “Perluasan Spektrum Fenotip Mutasi PAX2”Hu dkk. (2024) melaporkan identifikasi mutasi pergeseran kerangka c.76delG pada PAX2 dalam sebuah keluarga dengan glomerulosklerosis segmental fokal (FSGS) 1). Spektrum fenotip sindrom koloboma ginjal lebih luas dari yang diperkirakan sebelumnya, dan pentingnya skrining fungsi ginjal pada kasus koloboma diskus optikus kembali ditekankan. Disarankan untuk memperluas indikasi evaluasi fungsi ginjal pada pasien dengan koloboma.
FZD5 dan Abnormalitas Sinyal Wnt
Section titled “FZD5 dan Abnormalitas Sinyal Wnt”Cortes-Gonzalez dkk. (2024) melaporkan bahwa mutasi missense homozigot pada FZD5 (p.M160V) menyebabkan koloboma okular simptomatik dan mikrokornea 2). Analisis fungsional mengonfirmasi bahwa pola pewarisan resesif mengganggu aktivasi sinyal Wnt yang bergantung pada ligan. Tingkat diagnosis genetik koloboma kurang dari 30%, dan identifikasi gen penyebab baru diharapkan dapat berkontribusi pada peningkatan diagnosis.
Teknik Bedah Baru untuk Ablasi Retina
Section titled “Teknik Bedah Baru untuk Ablasi Retina”Jain dkk. (2024) melaporkan pelaksanaan reposisi retina dengan lem fibrin untuk ablasi retina terkait koloboma, dengan perbaikan visus akhir menjadi 20/50 3). Teknik pengolesan lem fibrin di sekitar robekan retina pada tepi koloboma untuk memperkuat adhesi merupakan pilihan tambahan yang menjanjikan untuk vitrektomi konvensional dengan fotokoagulasi. Akumulasi jumlah kasus dan verifikasi hasil jangka panjang merupakan tantangan di masa depan.
8. Referensi
Section titled “8. Referensi”-
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to FSGS. Mol Genet Genomic Med. 2024;12:e70006.
-
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in FZD5 causes syndromic ocular coloboma with microcornea. Hum Genet. 2024;143:1509-1521.
-
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
-
Pang CP, Lam DS. Differential occurrence of mutations causative of eye anomalies in families and sporadic patients with ocular coloboma. Hum Mutat. 2005;25(4):330.
-
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-194.
-
Chang L, Blain D, Bertuzzi S, Brooks BP. Uveal coloboma: clinical and basic science update. Curr Opin Ophthalmol. 2006;17(5):447-470.
-
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.