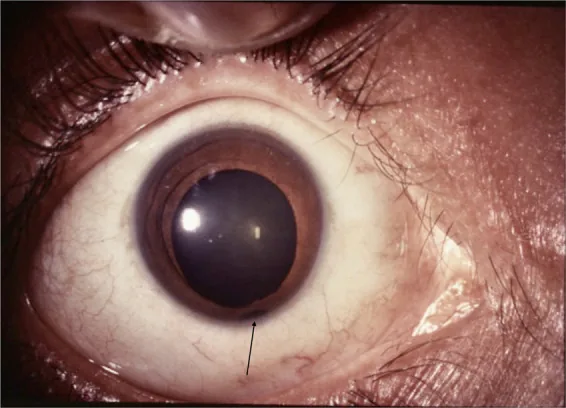
Iris
Pupil berbentuk lubang kunci: Defek biasanya terletak di kuadran inferonasal, menyebabkan pupil berubah bentuk seperti lubang kunci.
Lokasi inferotemporal: Kadang terjadi di posisi yang tidak khas.

Koloboma berasal dari bahasa Yunani yang berarti “cacat”, merupakan kelainan kongenital akibat gagal menutupnya celah embrional, menyebabkan defek jaringan di berbagai bagian mata. Dapat terjadi pada kelopak mata, iris, lensa, badan siliaris, koroid, retina, dan saraf optik. Defek biasanya terletak di kuadran inferonasal dan sering disertai mikrofthalmus.
Prevalensi diperkirakan 0,5–2,2 per 10.000 kelahiran. Di AS sekitar 2,6 per 10.000 kelahiran 4), di Eropa 4–19 per 100.000 kelahiran 6). Menyebabkan sekitar 11% kebutaan pada anak, dengan tingkat diagnosis genetik kurang dari 30% 6). Prevalensi koloboma kelopak mata 0,2–0,8 per 10.000 kelahiran. Terhitung 0,07% dari kelainan mata kongenital, dan 3,2–11,2% pada anak dengan gangguan penglihatan.
Koloboma dibedakan menjadi koloboma tipikal dan atipikal. Koloboma tipikal akibat gagal menutupnya celah embrional, terletak di inferonasal, sedangkan koloboma atipikal terjadi di lokasi lain dengan mekanisme perkembangan yang berbeda.
Kode ICD-10: Q10.3 (kelopak mata), Q13.0 (iris), Q12.2 (lensa), H47.319 (saraf optik), Q14.8 (koroid dan retina).
Ada yang sporadis dan herediter. Berbagai pola pewarisan telah dilaporkan, termasuk autosomal dominan, autosomal resesif, dan terkait-X. Beberapa gen penyebab telah diidentifikasi seperti PAX2, CHD7, dan FZD5, namun tingkat diagnosis genetik kurang dari 30% 6). Konseling genetik dianjurkan jika ada riwayat keluarga.
Ketajaman penglihatan sangat bervariasi, dari tidak ada persepsi cahaya hingga normal, tergantung pada lokasi dan luas defek.
Koloboma menunjukkan temuan khas di setiap bagian mata.
Iris
Pupil berbentuk lubang kunci: Defek biasanya terletak di kuadran inferonasal, menyebabkan pupil berubah bentuk seperti lubang kunci.
Lokasi inferotemporal: Kadang terjadi di posisi yang tidak khas.
Koroid dan Retina
Lesi putih kekuningan: Defek berbentuk bulat hingga seperti kipas dengan batas tegas di mana sklera tampak tembus.
Risiko ablasi retina: Insidensi 23–40%7). Pemantauan rutin diperlukan.
Saraf Optik dan Lensa
Pelebaran cekungan saraf optik: Mulai dari unilateral hingga bilateral, dengan derajat yang bervariasi.
Pendataran ekuator lensa: Terjadi akibat defek zonula Zinn. Diamati dalam keadaan midriasis.
Kelopak Mata
Defek medial kelopak mata atas: Defek jaringan penuh ketebalan.
Malformasi sistemik yang menyertai: Meskipun ada kasus terisolasi, seringkali disertai malformasi sistemik.
Koloboma iris jarang terjadi sendiri dan sering ditemukan berkesinambungan dengan koloboma koroid yang besar.
Ketajaman penglihatan bervariasi dari tidak ada persepsi cahaya hingga normal. Koloboma iris yang terbatas seringkali mempertahankan penglihatan. Jika melibatkan makula atau saraf optik, penglihatan cenderung buruk.
Penyebab utama koloboma adalah kegagalan penutupan fisura embrionik.
Fisura embrionik (fisura optik) terbentuk pada minggu ke-4 embrio dan selesai pada minggu ke-5. Penutupan dimulai pada minggu ke-6 dan selesai pada minggu ke-7. Jika proses penutupan ini terganggu karena suatu sebab, maka terjadilah koloboma. Peran vitamin A juga telah disebutkan.
Beberapa gen yang terlibat dalam terjadinya koloboma telah diidentifikasi.
| Gen | Penyakit/fenotipe terkait |
|---|---|
| PAX2 | Sindrom koloboma ginjal5) |
| CHD7 | Sindrom CHARGE |
| FZD5 | OC simptomatik + mikrokornea6) |
| TENM3 | MCOPS15 (mikrokornea + keterlambatan perkembangan)8) |
| FAT1 | Koloboma + nefropati9) |
| YAP1 | Terkait koloboma |
| ABCB6 | Terkait koloboma |
| SALL2 | Terkait koloboma |
Koloboma dapat menyertai sindrom sistemik berikut.
Sindrom CHARGE adalah sindrom malformasi multipel yang disebabkan oleh mutasi gen CHD7. Nama ini merupakan akronim dari Koloboma (C), Heart disease (H), Atresia koana (A), Retardasi pertumbuhan dan perkembangan (R), Genital hipoplasia (G), dan Ear abnormalities (E). Diagnosis ditegakkan berdasarkan kombinasi temuan-temuan tersebut.
Tes genetik komprehensif seperti whole exome sequencing (WES) dilakukan, namun tingkat diagnosis masih di bawah 30%6).
Koloboma perlu dibedakan dari penyakit berikut tergantung pada lokasinya.
| Lokasi | Diagnosis banding utama |
|---|---|
| Kelopak mata | Sindrom pita amnion, trauma |
| Iris | Aniridia, iridodialisis traumatik |
| Saraf optik | Sindrom morning glory, hipoplasia saraf optik |
Tidak ada pengobatan kuratif untuk koloboma; pengobatan berfokus pada terapi simtomatik dan manajemen komplikasi sesuai lokasi.
Castilla-Martinez dkk. (2024) melaporkan kasus koloboma iris, lensa, dan zonula Zinn yang disertai katarak, ditangani dengan femtosecond laser-assisted cataract surgery (FLACS) dan pupiloplasti, serta pemasangan CTR. Visus pascaoperasi membaik menjadi logMAR 0,24).
Pada koloboma saraf optik, terdapat displasia lamina kribrosa sehingga arteri dan vena sentralis retina sudah bercabang di belakang papil, dan pembuluh darah retina berasal dari beberapa titik di tepi papil. Di bawah papil sering ditemukan atrofi koroid dan retina akibat gagal menutupnya fisura embrional.
Untuk ablasioretina regmatogen, dilakukan vitrektomi. Teknik pembedahan seperti reposisi retina dengan lem fibrin7) dan fotokoagulasi endokular + tamponade gas3) telah dilaporkan. Pada ablasioretina serosa, beberapa kasus mengalami resolusi spontan, sehingga strategi pengobatan ditentukan secara individual.
Cawan optik terbentuk dari neuroektoderm pada minggu ke-4 embrio. Di sisi ventral cawan optik, terbentuk celah embrionik (celah cawan optik) yang dilalui oleh arteri hialoid. Celah ini selesai terbentuk pada minggu ke-5, dan penutupan dimulai pada minggu ke-6. Penutupan dimulai dari dekat ekuator dan berlanjut ke arah anterior (sisi iris) dan posterior (sisi saraf optik), selesai pada minggu ke-7.
Proses penutupan melibatkan transisi epitel-mesenkim (EMT). Sel-sel epitel retina saraf di tepi celah embrionik mendegradasi membran basal, memperoleh fenotip mesenkimal, dan menyatu. Gangguan pada proses ini menyebabkan koloboma.
Gen FZD5 mengode reseptor jalur sinyal Wnt. Mutasi kehilangan fungsi pada FZD5 menyebabkan abnormalitas sinyal Wnt, mengakibatkan kegagalan penutupan celah embrionik dan mikro kornea6).
Sel-sel krista neural (NCC) juga berperan dalam terjadinya koloboma. NCC berdiferensiasi menjadi jaringan mesenkim di sekitar cawan optik dan memainkan peran penting dalam proses penutupan celah embrionik2). Gangguan migrasi NCC menyebabkan kelainan perkembangan iris dan koroid.
Cortes-Gonzalez dkk. (2024) melaporkan bahwa mutasi missense homozigot pada FZD5 (p.M160V) menyebabkan koloboma okular simptomatik dan mikrokornea6). Mutasi ini menunjukkan pola pewarisan resesif, dan analisis fungsional mengonfirmasi bahwa aktivasi yang bergantung pada ligan dari jalur sinyal Wnt terganggu. Tingkat diagnosis genetik koloboma kurang dari 30%, dan identifikasi gen penyebab baru diharapkan dapat meningkatkan diagnosis.
Zhou dkk. (2022) melaporkan bahwa mutasi heterozigot majemuk pada gen TENM3 menyebabkan MCOPS15 (mikrokornea, koloboma iris-koroid, keterlambatan perkembangan umum)8). TENM3 mengkode protein transmembran yang terlibat dalam adhesi sel dan neurogenesis.
Esmaeilzadeh dkk. (2022) melaporkan bahwa mutasi gen FAT1 diidentifikasi pada sebuah keluarga Iran dengan koloboma iris dan nefropati9). FAT1 adalah anggota superfamili cadherin yang terlibat dalam polaritas sel dan morfogenesis jaringan.
Hu dkk. (2024) melaporkan bahwa mutasi pergeseran kerangka c.76delG pada PAX2 diidentifikasi pada sebuah keluarga dengan glomerulosklerosis segmental fokal (FSGS)5). Temuan ini menunjukkan bahwa spektrum fenotipe sindrom koloboma ginjal lebih luas dari yang diperkirakan sebelumnya.
Jain dkk. (2024) melaporkan satu kasus ablasi retina terkait koloboma yang menjalani reposisi retina dengan lem fibrin7). Teknik ini melibatkan pengolesan lem fibrin di sekitar robekan retina pada tepi koloboma untuk memperkuat adhesi, dan visus akhir membaik menjadi 20/50.
Ratra dkk. (2023) melaporkan keberhasilan penanganan kasus koloboma koroid atipikal dengan fistula sklera pasca-trauma menggunakan vitrektomi + fotokoagulasi endookular + tamponade gas3).
Scemla dkk. (2021) melaporkan kasus seorang pria berusia 19 tahun dengan filtrasi transskleral pada area koloboma koroid yang menyebabkan hipotoni (4 mmHg)1). Mikroskop ultrasonik biomikroskopi mengonfirmasi adanya defek sklera. Kondisi ini pulih secara spontan dalam 6 minggu, dengan tekanan intraokular 11 mmHg dan visus 1,0 yang tetap terjaga.