Aniridia Terisolasi
Frekuensi: Sekitar 2/3 dari total kasus.
Pola pewarisan: Autosomal dominan (AD).
Karakteristik: Disebabkan oleh mutasi gen PAX6. Tidak disertai gejala sistemik. Penetrasi lengkap, tetapi ekspresivitas bervariasi.

Aniridia adalah penyakit bawaan langka yang ditandai dengan hipoplasia atau defek iris dengan berbagai tingkat keparahan. Istilah “aniridia” sebenarnya keliru, karena fragmen jaringan iris hampir selalu dapat dideteksi dengan gonioskopi atau ultrasonografi biomikroskopi (UBM).
Prevalensi diperkirakan sekitar 1/40.000 hingga 1/100.000, tanpa perbedaan ras atau jenis kelamin yang signifikan 1). Dalam ICD-10, diklasifikasikan sebagai Q13.1.
Aniridia adalah penyakit pan-okular yang tidak hanya memengaruhi iris, tetapi juga kornea, lensa, sudut bilik mata, fovea, dan saraf optik 1), serta menimbulkan berbagai komplikasi okular yang mengancam penglihatan. Prognosis penglihatan umumnya buruk, dengan ketajaman penglihatan terkoreksi sekitar 0,1. Refleks pupil menghilang, namun refleks akomodasi masih ada, dan 60–90% kasus bersifat bilateral.
Tiga fenotipe berikut telah diidentifikasi.
Aniridia Terisolasi
Frekuensi: Sekitar 2/3 dari total kasus.
Pola pewarisan: Autosomal dominan (AD).
Karakteristik: Disebabkan oleh mutasi gen PAX6. Tidak disertai gejala sistemik. Penetrasi lengkap, tetapi ekspresivitas bervariasi.
Sindrom WAGR
Frekuensi: Sebagian dari kasus sporadis.
Pola pewarisan: Delesi berdekatan PAX6 dan WT1.
Karakteristik: Disertai tumor Wilms, kelainan urogenital, dan retardasi mental. Risiko tumor hingga 50%.
Sindrom Gillespie
Frekuensi: Sekitar 2% dari total kasus.
Pola pewarisan: Mutasi gen ITPR1.
Karakteristik: Disertai ataksia serebelar dan gangguan intelektual. Ditandai dengan kelainan iris spesifik berupa pupil yang tetap dan melebar 3).
Aniridia sporadis mencakup sekitar 1/3 dari total kasus dan disebabkan oleh delesi de novo pada 11p13 yang mencakup PAX6. Jika delesi meluas hingga gen WT1 yang berdekatan, hal ini menyebabkan sindrom WAGR 1). Sekitar 25–30% kasus aniridia sporadis berkembang menjadi tumor Wilms, dengan risiko relatif dilaporkan sebesar 67.
PAX6 adalah gen pengontrol utama pembentukan mata dan terlibat dalam perkembangan mata, tabung saraf, bulbus olfaktorius, pulau Langerhans pankreas, dan epitel olfaktorius. Penyakit ini timbul akibat hilangnya fungsi satu alel (haploinsufisiensi), dan kelainan pada kedua alel menyebabkan kematian janin. Pada tahun 2017, penyakit ini ditetapkan sebagai penyakit langka yang ditentukan oleh Undang-Undang Penyakit Langka, dan menjadi subjek subsidi biaya medis untuk tingkat keparahan III atau lebih tinggi (lihat bagian Diagnosis dan Pemeriksaan untuk detailnya) 7).
Kasus sporadis (mutasi baru) mencakup sekitar 1/3 dari seluruh kasus, dan dapat terjadi tanpa riwayat keluarga. Pada kasus sporadis, terdapat kemungkinan sindrom WAGR, sehingga pemeriksaan genetik dan skrining tumor Wilms dengan USG abdomen sangat penting.
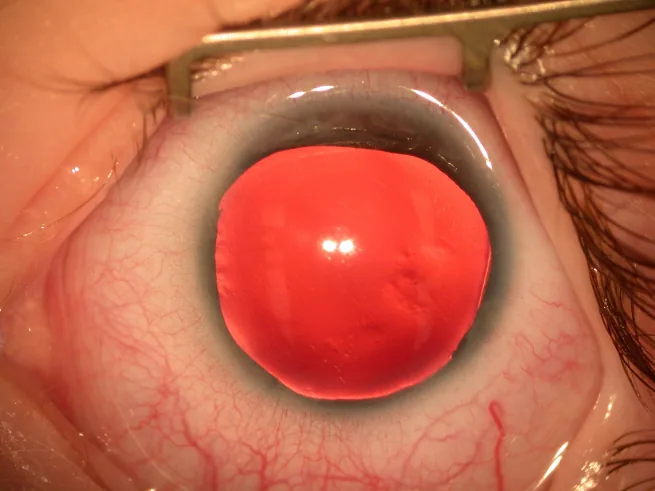
Sebagian besar kasus aniridia terdeteksi karena kelainan iris/pupil saat lahir, atau nistagmus pada masa bayi.
Fenotipe bervariasi antar dan dalam keluarga, namun perbedaan antara mata kanan dan kiri biasanya kecil.
Disebabkan terutama oleh hipoplasia fovea, ketajaman penglihatan terkoreksi seringkali sekitar 0,1-0,2. Prognosis penglihatan sangat buruk jika disertai hipoplasia makula. Koreksi refraksi sejak bayi dan perawatan low vision penting untuk perkembangan penglihatan.
PAX6 diekspresikan tidak hanya di jaringan mata tetapi juga di sistem saraf pusat, pulau Langerhans pankreas, dan epitel olfaktorius, sehingga komplikasi ekstraokular berikut dapat ditemukan 8).
Faktor penting yang menentukan fungsi penglihatan adalah glaukoma, hipoplasia makula, nistagmus, keratopati, katarak, dan kelainan pembentukan iris. Gangguan lapang pandang dan ketajaman penglihatan akibat glaukoma bersifat ireversibel, sehingga manajemen tekanan intraokular sangat penting dalam pemantauan.8)
Sebagian besar kasus aniridia kongenital disebabkan oleh mutasi heterozigot pada gen PAX6 yang terletak di lengan pendek kromosom 11 (11p13). Haploinsufisiensi PAX6 merupakan mekanisme utama terjadinya penyakit.1)
Gen PAX6 adalah gen master kontrol pembentukan mata dan berperan penting dalam perkembangan mata, tabung saraf, bulbus olfaktorius, dan pankreas. Perkembangan mata normal memerlukan dua salinan PAX6, dan kehilangan fungsi satu salinan saja sudah cukup untuk menyebabkan aniridia.1)
Studi kohort pada pasien Tiongkok mengidentifikasi mutasi penyebab pada gen PAX6 pada 96,9% kasus.1) Pada aniridia tipikal, mutasi yang menginduksi degradasi mRNA yang bergantung pada mutasi nonsense (NMD) atau delesi besar terdeteksi pada 96% kasus.1)
Secara patologis, otot polos iris tidak ada kecuali pada akar iris, dan terdapat disgenesis sudut bilik mata depan. Terdapat disfungsi sel punca epitel kornea, menyebabkan kelainan pada epitel dan membran Bowman, serta pembentukan pannus yang kaya pembuluh darah.
Rincian mutasi PAX6 yang menyebabkan fenotipe aniridia ditunjukkan di bawah ini.
| Tipe Mutasi | Frekuensi |
|---|---|
| Mutasi nonsense | Sekitar 39% |
| Mutasi pergeseran kerangka baca | Sekitar 25% |
| Mutasi sambungan | Sekitar 13% |
| Mutasi missense | Sekitar 12% |
Mutasi run-on (mutasi perpanjangan C-terminal) mencakup sekitar 5%, di mana kodon stop diubah menjadi kodon translasi sehingga menghasilkan protein PAX6 yang memanjang secara abnormal 6). Mutasi perpanjangan C-terminal sering disertai dengan hipoplasia iris yang berat dan gangguan penglihatan yang signifikan 1)6).
Mutasi gen yang paling sering adalah mutasi tipe PTC, dan mutasi missense juga telah dilaporkan 7). Mengenai kegunaan pengujian genetik, sekuensing Sanger atau NGS mendeteksi mutasi pada hampir 85% kasus aniridia terisolasi. Selain itu, MLPA atau CMA mendeteksi delesi dalam gen PAX6 atau daerah cis-regulator pada hampir 15% kasus 8).
Wang (2023) mengidentifikasi mutasi pergeseran kerangka baru c.640_646del (p.R214Pfs*28) dan melaporkan kasus dengan defek iris total, hipoplasia fovea, ektopia lentis, dan ablasi retina 1).
Ratna dkk. (2022) mengidentifikasi mutasi run-on c.1268A>T (p.*423L) pada sebuah keluarga India. Individu yang terkena menunjukkan aniridia total, nistagmus, hipoplasia fovea, AAK, subluksasi lensa ke atas, miopia tinggi, dan atrofi optik, yang menunjukkan fenotip berat akibat mutasi perpanjangan C-terminal 6).
Pada aniridia sporadis, delesi besar yang mencakup gen PAX6 dan WT1 menyebabkan sindrom WAGR. Risiko tumor Wilms pada kasus dengan delesi WT1 mencapai 50% 1). Jika dicurigai sindrom WAGR, pengujian genetik untuk mendeteksi delesi PAX6 dan WT1 memungkinkan penilaian risiko tumor Wilms dan pemantauan keterlambatan perkembangan 8). Evaluasi daerah WT1 melalui pengujian genetik sangat penting, dan 30% kasus sporadis dilaporkan mengembangkan tumor Wilms sebelum usia 5 tahun. Karena gen WT1 terletak di dekat PAX6, delesi lengan pendek kromosom 11 (delesi 11p13) yang menghilangkan kedua gen menyebabkan aniridia disertai tumor Wilms.
Sindrom Gillespie disebabkan oleh mutasi dominan negatif heterozigot atau mutasi bialelik pada gen ITPR1 3). Hingga saat ini, 37 kasus dengan diagnosis molekuler telah dilaporkan, dan residu Gly2554 dikenal sebagai titik panas 3).
Berdasarkan kriteria diagnosis aniridia (2020), diagnosis ditegakkan dengan kriteria berikut 7).
A. Gejala
B. Temuan Pemeriksaan
C. Diagnosis Banding
E. Pemeriksaan genetik: Mutasi patogenik pada gen PAX6 atau delesi pada wilayah 11p13
Kategori diagnosis7):
Klasifikasi tingkat keparahan untuk penetapan penyakit langka ditetapkan dalam 4 tingkatan berikut7).
| Tingkat Keparahan | Definisi |
|---|---|
| Derajat I | Satu mata terkena, mata lainnya normal |
| Derajat II | Kedua mata terkena, visus terkoreksi pada mata yang lebih baik ≥ 0,3 |
| Derajat III | Kedua mata terkena, visus terkoreksi pada mata yang lebih baik ≥ 0,1 dan < 0,3 |
| Derajat IV | Kedua mata terkena, visus terkoreksi pada mata yang lebih baik < 0,1 |
Bahkan pada derajat I–III, jika disertai penyempitan lapang pandang akibat glaukoma (sisa lapang pandang sentral ≤ 20° dengan target Goldmann I/4), maka derajat keparahan naik satu tingkat. Derajat keparahan ≥ III menjadi sasaran subsidi biaya medis 7).
Diagnosis klinis mudah ditegakkan dengan melihat defek atau hipoplasia iris menggunakan slit-lamp. Evaluasi sisa jaringan iris dilakukan dengan gonioskopi atau ultrasound biomicroscopy. Periksa juga ada tidaknya kelainan perkembangan sudut bilik mata depan.
Evaluasi secara sistematis komplikasi mata berikut:
Tujuan paling penting dalam evaluasi genetik aniridia adalah memastikan apakah delesi PAX6 meluas hingga gen WT11). Mutasi dan delesi pada daerah PAX6 dan WT1 dievaluasi menggunakan sekuensing eksom lengkap atau MLPA1)2).
Pada aniridia sporadis, evaluasi risiko tumor Wilms akibat delesi gen WT1 berkaitan langsung dengan prognosis hidup1). Meskipun kasus familial, karena variabilitas fenotipe, diagnosis pasti melalui tes genetik dan konseling genetik dianjurkan.
Tidak ada terapi kuratif untuk aniridia. Perawatan utama adalah perawatan low vision untuk memaksimalkan sisa penglihatan dan penanganan individual untuk setiap komplikasi8).
Transplantasi kornea untuk kekeruhan stroma kornea harus dipertimbangkan secara hati-hati8).
Transplantasi kornea dapat memberikan perbaikan penglihatan jangka pendek, namun perbaikannya terbatas karena komplikasi seperti hipoplasia makula. Dalam jangka panjang, prognosis penglihatan buruk akibat progresi glaukoma dan disfungsi graft.
Pada defisiensi sel punca epitel kornea, pertimbangkan perawatan bedah8).
Operasi katarak dipertimbangkan berdasarkan tingkat kekeruhan dan fotofobia8).
Katarak terjadi pada 50–85% kasus sebelum usia 20 tahun. Operasi direncanakan berdasarkan intensitas kekeruhan dan fotofobia. Dilaporkan bahwa 66–100% kasus operasi mengalami perbaikan penglihatan, namun perlu diperhatikan hal-hal berikut.
Karena zonula Zinn rapuh, pemasangan lensa intraokular harus dilakukan dengan indikasi yang hati-hati.
Hu dkk. (2024) melakukan fakoemulsifikasi dengan bantuan iluminasi balik tipe chandelier pada 2 kasus aniridia kongenital dengan AAK berat. Visualisasi intraoperatif biasa sulit karena kekeruhan kornea, namun iluminasi dari posterior memungkinkan visualisasi yang jelas dari lensa dan kapsul anterior, dan pada 3 minggu pascaoperasi, ketajaman visual terkoreksi membaik menjadi 20/200 dan 20/1000 masing-masing4).
Glaukoma secara langsung berkaitan dengan prognosis fungsi penglihatan, sehingga perlu ditangani secara aktif8).
Setelah timbul glaukoma, penanganan dilakukan berdasarkan algoritma 5 langkah berikut.
Terapi obat: β-blocker, stimulan simpatis, dan obat terkait prostaglandin (PG) efektif. Brimonidin (stimulan reseptor α-adrenergik) pada bayi memiliki risiko depresi sistem saraf pusat, sehingga kontraindikasi pada anak di bawah 2 tahun. Jika ada kekhawatiran kerusakan epitel kornea, gunakan sediaan tanpa pengawet.
Operasi rekonstruksi saluran keluar (goniotomi/trabekulotomi): Direkomendasikan sebagai operasi pertama16). Ada juga laporan mengenai goniotomi profilaksis. Namun, pada kasus di mana sisa iris menutupi trabekulum, prosedur ini mungkin tidak efektif.
Operasi filtrasi (trabekulektomi): Hanya terbatas pada laporan jangka pendek dan menengah dengan jumlah kasus kecil. Pada mata anak-anak, cenderung memberikan hasil yang buruk, dan fistula bola mata pascaoperasi mencapai sekitar 25%13). Ada juga laporan glaukoma maligna pascaoperasi.
Operasi implan glaukoma (operasi shunt tuba): Perangkat tipe Baerveldt dan Ahmed dapat digunakan. Pemasangan tuba pada mata dengan lensa alami direkomendasikan secara tangensial, bukan ke arah tengah kornea. Kontrol tekanan intraokular yang baik dapat diharapkan.
Koagulasi badan siliaris: pilihan terakhir. Dilaporkan bahwa banyak kasus kriokoagulasi badan siliaris berakhir dengan atrofi bola mata. Karena terdapat hipoplasia badan siliaris, risiko atrofi bola mata lebih tinggi dibandingkan mata normal.
Karena adanya kelainan perkembangan sudut bilik mata, diperlukan pendekatan yang berbeda dari glaukoma sudut terbuka biasa. Pilihan pertama adalah operasi rekonstruksi saluran keluar, kemudian operasi shunt tube menjadi pilihan yang baik. Brimonidine dikontraindikasikan pada anak di bawah 2 tahun, dan penggunaan obat antimetabolit dapat memperburuk AAK sehingga diperlukan pertimbangan yang hati-hati 8).
Perawatan low vision harus dimulai sejak dini8).
Koreksi refraksi adalah dasar, dengan tingkat komplikasi miopia lebih dari 64%.
Penanganan fotofobia penting untuk menjaga perkembangan fungsi penglihatan dan kualitas hidup8).
Sebagian besar pasien dapat bersekolah di kelas reguler, tetapi memerlukan dukungan seperti buku teks cetak besar, tablet, dan rak buku. Pilihan lainnya termasuk mengikuti kelas tunanetra atau memanfaatkan konsultasi pengasuhan dan pendidikan dari sekolah tunanetra atau sekolah dukungan penglihatan khusus.
Sejak April 2017, kondisi ini telah ditetapkan sebagai penyakit langka yang ditunjuk, sehingga meskipun belum memiliki kartu disabilitas fisik, pasien dengan tingkat keparahan III atau lebih berhak mendapatkan bantuan biaya medis dan penyediaan alat bantu 7). Alat bantu yang termasuk meliputi kacamata korektif, kacamata pelindung sinar, lensa kontak (termasuk yang dilengkapi iris buatan), kacamata low vision, tongkat keselamatan untuk tunanetra, dan mata palsu.
PAX6 mencakup 22 kb DNA genom yang mengandung 14 ekson dan mengkode 422 asam amino 1). Gen ini memiliki dua domain pengikat DNA (paired domain dan paired-type homeodomain), dan domain PST (kaya prolin, serin, treonin) di ujung C berfungsi sebagai aktivator transkripsi.
PAX6 mengatur proliferasi, diferensiasi, migrasi, dan adhesi sel, dan targetnya mencakup PAX6 itu sendiri serta gen yang mengkode kristalin lensa dan keratin kornea. Ekspresinya berlanjut di retina, lensa, dan kornea orang dewasa. Gen PAX6 adalah salah satu gen master kontrol yang mengatur diferensiasi organ selama masa embrio.
Sebagian besar mutasi PAX6 menyebabkan haploinsufisiensi melalui mekanisme nonsense-mediated mRNA decay (NMD) 1). Mutasi yang memperkenalkan kodon stop prematur (PTC) (mutasi nonsense, mutasi pergeseran bingkai, dan sebagian besar mutasi sambungan) menghasilkan fenotip aniridia tipikal.
Di sisi lain, jika PTC terletak di ekson terakhir atau dalam 50 bp terakhir dari ujung ekson kedua terakhir, mutasi tersebut dapat lolos dari NMD, dan protein terpotong dapat diterjemahkan, berpotensi menyebabkan fenotip yang lebih parah 1).
Kasus langka telah dilaporkan di mana mutasi nonsense PAX6 c.282C>A (p.Cys94*) dan trisomi 21 terjadi bersamaan pada pasien yang sama. Mutasi PAX6 terjadi secara de novo, menyebabkan aniridia bilateral lengkap, glaukoma kongenital, AAK, dan hipoplasia fovea 2).
Meskipun korelasi genotip-fenotip yang jelas belum ditetapkan, beberapa kecenderungan diketahui 1).
Seri gonioskopi Grant dan Walton menunjukkan bahwa pada tahap awal, stroma iris meluas ke anterior di atas trabekula membentuk perlengketan seperti sinekia, yang secara bertahap menjadi seperti lembaran dan akhirnya menyebabkan oklusi sudut14). Mekanisme ini merupakan faktor utama terjadinya glaukoma. Secara patologis, dasar dari kondisi ini adalah defek otot polos yang menyisakan akar iris dan disgenesis sudut bilik mata depan.
AAK terutama disebabkan oleh defisiensi sel punca limbal (LSCD), namun juga melibatkan diferensiasi abnormal epitel kornea, gangguan adhesi, invasi sel konjungtiva, dan produksi air mata yang tidak mencukupi. Defisiensi matriks metaloproteinase-9 (MMP-9) yang diatur oleh PAX6 menyebabkan akumulasi fibrin dan infiltrasi sel inflamasi, yang mengakibatkan hilangnya transparansi akibat gangguan susunan kolagen stroma.
AAK diklasifikasikan menjadi 5 tahap. Tahap I hanya menunjukkan kelainan epitel perifer, Tahap II menunjukkan perubahan epitel sentripetal (belum mencapai pusat), Tahap III menunjukkan perubahan epitel kornea sentral dan neovaskularisasi superfisial perifer, Tahap IV menunjukkan neovaskularisasi superfisial seluruh kornea, dan Tahap V menunjukkan kelainan epitel seluruh kornea dan sikatrik stroma dalam10).
Terdapat hubungan antara status mutasi PAX6 dan perkembangan AAK. Pada pasien dengan mutasi PTC atau perpanjangan C-terminal, AAK berkembang secara bergantung pada usia, sedangkan pada tipe mutasi lain dapat terjadi keratopati non-progresif11).
Sindrom Gillespie disebabkan oleh mutasi pada gen ITPR13). ITPR1 merupakan anggota famili reseptor IP3 yang membentuk saluran pelepasan Ca²⁺ dan terlokalisasi di retikulum endoplasma. Mutasi dominan negatif memengaruhi pembentukan dan pemeliharaan sfingter iris, menyebabkan hipoplasia iris spesifik di sekitar pupil dan midriasis fiks.
Dalam tinjauan literatur sindrom Gillespie oleh Ciaccio dkk. (2024), analisis terhadap 33 kasus yang terkonfirmasi secara molekuler menunjukkan bahwa perkembangan motorik tertunda namun membaik seiring waktu, keterbelakangan intelektual tidak ditemukan pada semua kasus (17% memiliki inteligensi normal), dan tanda neurologis bersifat non-progresif3).
Dengan meluasnya teknologi sekuensing eksom utuh, identifikasi mutasi PAX6 baru terus berlanjut. Pada tahun 2018, 491 mutasi telah terdaftar di Human PAX6 Mutation Database, dan sekitar 250 mutasi baru telah dilaporkan sejak saat itu 1). Mutasi pada daerah nonkoding juga mulai diidentifikasi sebagai penyebab aniridia, yang diharapkan dapat menjelaskan kasus yang sebelumnya tidak terdiagnosis dengan pemeriksaan konvensional 9).
Pada operasi katarak pada kasus dengan AAK berat, teknik visualisasi dengan bantuan iluminasi balik chandelier berguna 4). Teknik ini memungkinkan fakoemulsifikasi yang aman bahkan pada pasien dengan AAK derajat 3–4, dan perbaikan visus pascaoperasi telah tercapai.
Telah diketahui bahwa pola progresi AAK berbeda tergantung pada jenis mutasi PAX6. Dengan menurunnya biaya tes genetik, prediksi perjalanan klinis berdasarkan tipe mutasi dan intervensi dini menjadi pilihan yang realistis.
Pada kasus gabungan aniridia dan trisomi 21, telah dilaporkan contoh dengan perjalanan yang relatif ringan meskipun kedua penyakit tersebut ada 2). Memahami dampak pada fenotipe ketika beberapa kelainan genetik ada pada pasien yang sama dapat memberikan wawasan penting untuk pengobatan personal di masa depan.
Penerapan obat readthrough (ataluren) untuk mutasi tipe PTC pada aniridia sedang diteliti pada tingkat penelitian dasar 8). Mengenai terapi gen PAX6, penelitian dasar tentang pengisian gen menggunakan vektor AAV-PAX6 pada model tikus mutan Sey sedang berlangsung. Diharapkan dapat dikembangkan ke uji klinis di masa depan.
Uji coba transplantasi lembaran sel epitel kornea yang berasal dari sel iPS sedang dilakukan di dalam dan luar negeri, dan menarik perhatian sebagai terapi baru untuk AAK 8). Iris buatan (seperti CustomFlex Artificial Iris) telah memiliki akumulasi pengalaman penggunaan di luar negeri. Lensa kontak dengan iris buatan sebagai alat bantu termasuk dalam cakupan asuransi.
Akumulasi data registri skala besar di Jepang untuk memahami kondisi aktual dan peningkatan kualitas bukti merupakan isu penting di masa depan 8). Prediksi progresi AAK berdasarkan mutasi gen individu dan optimalisasi intervensi dini diharapkan dapat tercapai.