ภาวะไม่มีม่านตาแบบแยกเดี่ยว
ความถี่: ประมาณ 2 ใน 3 ของทั้งหมด
รูปแบบการถ่ายทอดทางพันธุกรรม: ลักษณะเด่นบนออโตโซม (AD)
ลักษณะเด่น: เกิดจากการกลายพันธุ์ของยีน PAX6 ไม่มีอาการทางระบบอื่น การแทรกซึมสมบูรณ์แต่การแสดงออกหลากหลาย

ภาวะไม่มีม่านตา (Aniridia) เป็นโรคประจำตัวที่พบได้ยาก ซึ่งมีลักษณะเฉพาะคือม่านตามีการเจริญไม่สมบูรณ์หรือขาดหายไปในระดับต่างๆ ชื่อ “ภาวะไม่มีม่านตา” เป็นการเรียกที่ไม่ถูกต้องนัก เนื่องจากการตรวจด้วย gonioscopy หรือ ultrasound biomicroscopy (UBM) มักจะพบเศษเนื้อเยื่อม่านตาเกือบตลอดเวลา
ความชุกของโรคอยู่ที่ประมาณ 1 ใน 40,000 ถึง 1 ใน 100,000 ราย และไม่พบความแตกต่างอย่างมีนัยสำคัญทางเชื้อชาติหรือเพศ1) ในรหัส ICD-10 จัดอยู่ในกลุ่ม Q13.1
โรคนี้เป็นโรคที่ส่งผลกระทบต่อลูกตาทั้งหมด ไม่เพียงแต่ม่านตาเท่านั้น แต่ยังรวมถึงกระจกตา เลนส์แก้วตา มุมลูกตา รอยบุ๋มจอตา และเส้นประสาทตา1) และทำให้เกิดภาวะแทรกซ้อนทางตาหลายอย่างที่คุกคามการมองเห็น การพยากรณ์โรคด้านการมองเห็นโดยทั่วไปไม่ดี โดยมักมีค่าสายตาที่แก้ไขแล้วอยู่ที่ประมาณ 0.1 ปฏิกิริยารูม่านตาหายไป แต่ปฏิกิริยาการปรับโฟกัสยังคงอยู่ และ 60-90% เป็นโรคที่เกิดกับตาทั้งสองข้าง
พบฟีโนไทป์ 3 แบบดังนี้
ภาวะไม่มีม่านตาแบบแยกเดี่ยว
ความถี่: ประมาณ 2 ใน 3 ของทั้งหมด
รูปแบบการถ่ายทอดทางพันธุกรรม: ลักษณะเด่นบนออโตโซม (AD)
ลักษณะเด่น: เกิดจากการกลายพันธุ์ของยีน PAX6 ไม่มีอาการทางระบบอื่น การแทรกซึมสมบูรณ์แต่การแสดงออกหลากหลาย
กลุ่มอาการ WAGR
ความถี่: พบในบางส่วนของผู้ป่วยที่เกิดขึ้นเอง
รูปแบบการถ่ายทอดทางพันธุกรรม: การขาดหายของยีน PAX6 และ WT1 ที่อยู่ติดกัน
ลักษณะเด่น: ร่วมกับเนื้องอก Wilms, ความผิดปกติของระบบสืบพันธุ์และทางเดินปัสสาวะ, และภาวะบกพร่องทางสติปัญญา ความเสี่ยงในการเกิดเนื้องอกสูงถึง 50%
กลุ่มอาการกิลเลสปี
ความถี่: ประมาณ 2% ของทั้งหมด
รูปแบบการถ่ายทอดทางพันธุกรรม: การกลายพันธุ์ของยีน ITPR1
ลักษณะเด่น: ร่วมกับภาวะสมองน้อยเสื่อมและความบกพร่องทางสติปัญญา ลักษณะเฉพาะคือความผิดปกติของม่านตาที่ทำให้รูม่านตาขยายคงที่ 3)
ภาวะไม่มีม่านตาที่เกิดขึ้นเป็นครั้งคราวคิดเป็นประมาณ 1 ใน 3 ของทั้งหมด เกิดจากการขาดหายของยีน 11p13 ซึ่งรวมถึง PAX6 แบบ de novo หากการขาดหายขยายไปถึงยีน WT1 ที่อยู่ติดกัน จะทำให้เกิด WAGR syndrome 1) ผู้ป่วยภาวะไม่มีม่านตาที่เกิดขึ้นเป็นครั้งคราวร้อยละ 25-30 จะเกิด Wilms tumor และรายงานความเสี่ยงสัมพัทธ์เท่ากับ 67
PAX6 เป็นยีนควบคุมหลักในการสร้างดวงตา เกี่ยวข้องกับการพัฒนาของดวงตา ท่อประสาท ปุ่มรับกลิ่น เกาะ Langerhans ในตับอ่อน และเยื่อบุรับกลิ่น การสูญเสียการทำงานของอัลลีลหนึ่ง (haploinsufficiency) ทำให้เกิดโรค หากอัลลีลทั้งสองผิดปกติจะทำให้ทารกในครรภ์เสียชีวิต ในปี 2017 โรคนี้ถูกกำหนดให้เป็นโรคหายากตามกฎหมายโรคหายาก และผู้ที่มีความรุนแรงระดับ III ขึ้นไป (ดูรายละเอียดในหัวข้อการวินิจฉัยและการตรวจ) จะมีสิทธิ์ได้รับการช่วยเหลือค่าใช้จ่ายทางการแพทย์ 7)
กรณีที่เกิดขึ้นเอง (การกลายพันธุ์ใหม่) คิดเป็นประมาณ 1 ใน 3 ของผู้ป่วยทั้งหมด และสามารถเกิดได้แม้ไม่มีประวัติครอบครัว ในกรณีที่เกิดขึ้นเองอาจมีความเป็นไปได้ของ WAGR syndrome ดังนั้นการตรวจทางพันธุกรรมและการตรวจอัลตราซาวนด์ช่องท้องเพื่อคัดกรอง Wilms tumor จึงมีความสำคัญ
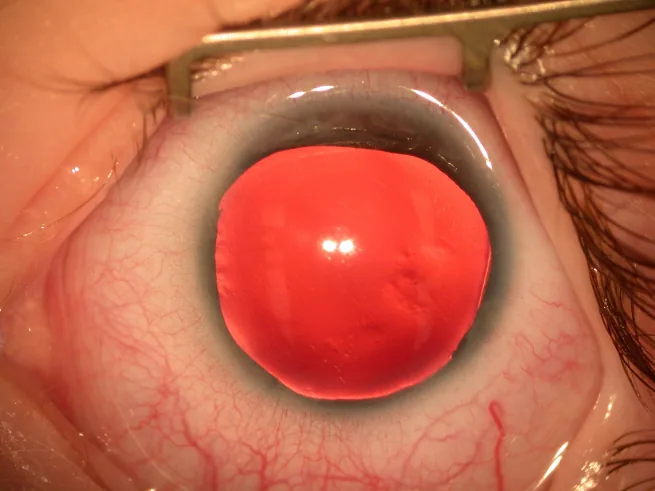
ภาวะไม่มีม่านตามักถูกตรวจพบจากความผิดปกติของม่านตาหรือรูม่านตาตั้งแต่แรกเกิด หรือจากอาการตากระตุกในวัยทารก
ลักษณะทางฟีโนไทป์แตกต่างกันระหว่างครอบครัวและภายในครอบครัวเดียวกัน แต่ความแตกต่างระหว่างตาซ้ายและตาขวามักมีน้อย
สาเหตุหลักคือการสร้างรอยบุ๋มจอตาที่ไม่สมบูรณ์ ทำให้สายตาที่แก้ไขแล้วมักอยู่ที่ประมาณ 0.1–0.2 หากมีภาวะจอตาส่วนกลางเจริญไม่เต็มที่ร่วมด้วย การพยากรณ์โรคทางสายตาจะแย่เป็นพิเศษ การแก้ไขค่าสายตาผิดปกติและการดูแลสายตาเลือนรางตั้งแต่ทารกมีความสำคัญต่อพัฒนาการทางสายตา
เนื่องจาก PAX6 แสดงออกในเนื้อเยื่อตา รวมถึงระบบประสาทส่วนกลาง เซลล์เกาะเล็กแลงเกอร์ฮานส์ในตับอ่อน และเยื่อบุรับกลิ่น จึงอาจพบภาวะแทรกซ้อนนอกตาต่อไปนี้8)
ปัจจัยสำคัญที่กำหนดการมองเห็น ได้แก่ ต้อหิน, จุดภาพชัดเจริญผิดปกติ, อาตา, โรคกระจกตา, ต้อกระจก, และความผิดปกติของม่านตา ความเสียหายของลานสายตาและการมองเห็นจากต้อหินนั้นไม่สามารถกลับคืนได้ ดังนั้นการจัดการความดันลูกตาจึงสำคัญที่สุดในการติดตามผล8)
ภาวะไม่มีม่านตาแต่กำเนิดส่วนใหญ่เกิดจากการกลายพันธุ์แบบเฮเทอโรไซกัสของยีน PAX6 ซึ่งอยู่บนแขนสั้นของโครโมโซมคู่ที่ 11 (11p13) กลไกหลักของการเกิดโรคคือ haploinsufficiency ของ PAX61)
ยีน PAX6 เป็นยีนควบคุมหลักในการสร้างดวงตา และมีบทบาทสำคัญในการพัฒนาของดวงตา, ท่อประสาท, ปุ่มรับกลิ่น, และตับอ่อน การพัฒนาดวงตาตามปกติต้องใช้ PAX6 จำนวน 2 copies และการสูญเสียการทำงานของเพียง 1 copy ก็สามารถทำให้เกิดภาวะไม่มีม่านตาได้1)
ในการศึกษาแบบ cohort ในผู้ป่วยชาวจีน พบการกลายพันธุ์ที่เป็นสาเหตุในยีน PAX6 ใน 96.9% ของผู้ป่วย1) ในภาวะ aniridia ทั่วไป การกลายพันธุ์ที่ทำให้เกิด nonsense-mediated mRNA decay (NMD) หรือการขาดหายขนาดใหญ่ตรวจพบได้ใน 96%1)
ทางพยาธิวิทยา พบว่ากล้ามเนื้อเรียบหายไป ยกเว้นบริเวณโคนม่านตา และพบความผิดปกติของมุมลูกตา การทำงานของเซลล์ต้นกำเนิดเยื่อบุผิวกระจกตาผิดปกติ ทำให้เกิดความผิดปกติของเยื่อบุผิวและเยื่อ Bowman และเกิด pannus ที่มีหลอดเลือดมาก
รายละเอียดของการกลายพันธุ์ของ PAX6 ที่ทำให้เกิดฟีโนไทป์ของ aniridia แสดงดังนี้
| ประเภทการกลายพันธุ์ | ความถี่ |
|---|---|
| การกลายพันธุ์แบบ nonsense | ประมาณ 39% |
| การกลายพันธุ์แบบ frameshift | ประมาณ 25% |
| การกลายพันธุ์แบบ splice | ประมาณ 13% |
| การกลายพันธุ์แบบมิสเซนส์ | ประมาณ 12% |
การกลายพันธุ์แบบรันออน (การกลายพันธุ์แบบต่อปลาย C) คิดเป็นประมาณ 5% เกิดจากโคดอนหยุดถูกแปลงเป็นโคดอนแปลรหัส ทำให้เกิดโปรตีน PAX6 ที่ยาวผิดปกติ 6) การกลายพันธุ์แบบต่อปลาย C มักเกี่ยวข้องกับภาวะม่านตาพร่องรุนแรงและความบกพร่องทางการมองเห็นระดับสูง 1)6)
การกลายพันธุ์ทางพันธุกรรมส่วนใหญ่เป็นชนิด PTC และยังมีรายงานการกลายพันธุ์แบบมิสเซนส์ 7) เกี่ยวกับประโยชน์ของการตรวจทางพันธุกรรม การหาลำดับดีเอ็นเอแบบแซงเจอร์หรือ NGS สามารถตรวจพบการกลายพันธุ์ในผู้ป่วย isolated aniridia ได้เกือบ 85% นอกจากนี้ MLPA หรือ CMA สามารถตรวจพบการขาดหายภายในยีน PAX6 หรือบริเวณควบคุมซิสในผู้ป่วยเกือบ 15% 8)
Wang (2023) ระบุการกลายพันธุ์แบบเฟรมชิฟต์ c.640_646del (p.R214Pfs*28) ใหม่ และรายงานผู้ป่วยที่มีภาวะม่านตาขาดทั้งหมด ฟอฟเวียเจริญไม่เต็มที่ เลนส์แก้วตาเคลื่อน และจอประสาทตาลอก 1)
Ratna และคณะ (2022) ระบุการกลายพันธุ์แบบ run-on c.1268A>T (p.*423L) ในครอบครัวชาวอินเดีย ผู้ป่วยมีอาการ aniridia สมบูรณ์ อาตา foveal hypoplasia AAK เลนส์เคลื่อนขึ้นด้านบน สายตาสั้นรุนแรง และ optic atrophy ซึ่งแสดงฟีโนไทป์รุนแรงจากการกลายพันธุ์ที่ยืดปลาย C 6)
ใน aniridia ชนิดประปราย การขาดหายขนาดใหญ่ที่รวมยีน PAX6 และ WT1 เป็นสาเหตุของ WAGR syndrome ความเสี่ยงของ Wilms tumor เมื่อมีการขาด WT1 สูงถึง 50% 1) หากสงสัย WAGR syndrome ควรตรวจทางพันธุกรรมเพื่อยืนยันการขาด PAX6 และ WT1 ซึ่งจะช่วยประเมินความเสี่ยง Wilms tumor และติดตามพัฒนาการล่าช้า 8) การประเมินบริเวณ WT1 ด้วยการตรวจยีนเป็นสิ่งจำเป็น โดย 30% ของผู้ป่วยประปรายจะเกิด Wilms tumor ก่อนอายุ 5 ปี เนื่องจากยีน WT1 อยู่ใกล้กับ PAX6 การขาดโครโมโซม 11 แขนสั้น (11p13 deletion) ที่ทำให้ทั้งสองยีนขาดหาย จึงทำให้ aniridia ร่วมกับ Wilms tumor
กลุ่มอาการกิลเลสปีเกิดจากการกลายพันธุ์แบบเฮเทอโรไซกัสเด่นเชิงลบหรือการกลายพันธุ์แบบสองอัลลีลในยีน ITPR1 3) มีรายงานผู้ป่วยที่ได้รับการวินิจฉัยระดับโมเลกุลแล้ว 37 ราย โดยกรดอะมิโน Gly2554 เป็นจุดร้อนที่ทราบกันดี 3)
การวินิจฉัยภาวะไม่มีม่านตาเป็นไปตามเกณฑ์การวินิจฉัย (พ.ศ. 2563) โดยยืนยันตามเกณฑ์ต่อไปนี้7)
ก. อาการ
B. ผลการตรวจทางห้องปฏิบัติการ
C. โรคที่ควรแยกวินิจฉัย
E. การตรวจทางพันธุกรรม: การกลายพันธุ์ที่ก่อโรคของยีน PAX6 หรือการขาดหายของบริเวณ 11p13
ประเภทการวินิจฉัย7):
การจำแนกความรุนแรงสำหรับการรับรองโรคหายากถูกกำหนดไว้เป็น 4 ระดับดังต่อไปนี้7)
| ความรุนแรง | คำจำกัดความ |
|---|---|
| ระดับ I | ตาข้างหนึ่งเป็นโรค อีกข้างปกติ |
| ระดับ II | เป็นโรคทั้งสองข้าง ค่าสายตาที่แก้ไขแล้วของข้างที่ดีกว่าอยู่ที่ 0.3 ขึ้นไป |
| ระดับ III | เป็นโรคทั้งสองข้าง ค่าสายตาที่แก้ไขแล้วของข้างที่ดีกว่าอยู่ที่ 0.1 ขึ้นไปแต่ไม่ถึง 0.3 |
| ระดับ IV | เป็นโรคตาทั้งสองข้าง ค่าความคมชัดของสายตาที่แก้ไขแล้วในตาข้างที่ดีน้อยกว่า 0.1 |
แม้จะเป็นระดับ I–III หากมีภาวะลานสายตาแคบจากโรคต้อหินหรืออื่นๆ (ลานสายตาส่วนกลางเหลือน้อยกว่า 20 องศาโดยใช้ Goldmann I/4) จะเลื่อนไปยังระดับความรุนแรงที่สูงขึ้นหนึ่งระดับ ระดับความรุนแรง III ขึ้นไปมีสิทธิ์ได้รับการช่วยเหลือค่ารักษาพยาบาล7)
การวินิจฉัยทางคลินิกทำได้ง่ายโดยใช้กล้องจุลทรรศน์ชนิดร่องกราด (slit-lamp microscope) เพื่อตรวจหาการขาดหายหรือการเจริญไม่สมบูรณ์ของม่านตา การประเมินเนื้อเยื่อม่านตาที่เหลือทำได้โดย gonioscopy หรือ ultrasound biomicroscopy นอกจากนี้ยังต้องตรวจสอบความผิดปกติของการพัฒนาของมุมลูกตาส่วนหน้าด้วย
ประเมินภาวะแทรกซ้อนทางตาต่อไปนี้อย่างเป็นระบบ:
เป้าหมายที่สำคัญที่สุดในการประเมินทางพันธุกรรมของภาวะ aniridia คือการยืนยันว่าการขาดหายของ PAX6 ครอบคลุมไปถึงยีน WT1 หรือไม่ 1) การประเมินการกลายพันธุ์และการขาดหายในบริเวณ PAX6 และ WT1 ทำได้โดยการวิเคราะห์ exome ทั้งหมดหรือวิธี MLPA 1)2)
ในภาวะ aniridia ที่เกิดขึ้นเป็นครั้งคราว การประเมินความเสี่ยงของเนื้องอก Wilms จากการขาดหายของยีน WT1 ส่งผลโดยตรงต่อการพยากรณ์โรค1) แม้ในกรณีที่เป็น familial ก็มีความหลากหลายของฟีโนไทป์ ดังนั้นจึงแนะนำให้ตรวจทางพันธุกรรมเพื่อการวินิจฉัยที่แน่นอนและการให้คำปรึกษาทางพันธุกรรม
ไม่มีวิธีการรักษาที่เป็นสาเหตุของภาวะไม่มีม่านตา (aniridia) การดูแลการมองเห็นเลือนราง (low vision care) เพื่อใช้การมองเห็นที่เหลืออยู่ให้เกิดประโยชน์สูงสุด และการรักษาเฉพาะสำหรับภาวะแทรกซ้อนแต่ละอย่างเป็นหลักในการจัดการ8)
ควรพิจารณาการปลูกถ่ายกระจกตาสำหรับภาวะขุ่นของเนื้อกระจกตาอย่างรอบคอบ8)
การปลูกถ่ายกระจกตาอาจช่วยให้การมองเห็นดีขึ้นในระยะสั้น แต่การปรับปรุงมีจำกัดเนื่องจากภาวะแทรกซ้อนร่วม เช่น ภาวะจอประสาทตาส่วนรับภาพเจริญผิดปกติ (macular hypoplasia) ในระยะยาว การดำเนินของโรคต้อหินและความล้มเหลวของ graft ส่งผลให้พยากรณ์โรคการมองเห็นไม่ดี
ในกรณีที่เซลล์ต้นกำเนิดเยื่อบุกระจกตาหมดสภาพ ควรพิจารณาการผ่าตัดรักษา8)
การผ่าตัดต้อกระจกจะพิจารณาจากระดับความขุ่นและอาการกลัวแสง8)
ต้อกระจกเกิดขึ้นใน 50–85% ของผู้ป่วยก่อนอายุ 20 ปี การผ่าตัดจะวางแผนตามความรุนแรงของความขุ่นและอาการกลัวแสง มีรายงานว่าผู้ป่วย 66–100% ที่ได้รับการผ่าตัดมีสายตาดีขึ้น แต่ต้องระวังประเด็นต่อไปนี้:
เนื่องจาก Zinn zonule มีความเปราะบาง การใส่เลนส์แก้วตาเทียมจึงต้องพิจารณาอย่างรอบคอบ
Hu และคณะ (2024) ได้ทำการผ่าตัดสลายต้อกระจกด้วยคลื่นเสียงความถี่สูงภายใต้การช่วยส่องสว่างจากด้านหลังแบบโคมระย้าในผู้ป่วยภาวะไม่มีม่านตาแต่กำเนิดที่มี AAK รุนแรงจำนวน 2 ราย แม้การมองเห็นระหว่างผ่าตัดตามปกติจะทำได้ยากเนื่องจากกระจกตาขุ่นมัว แต่การส่องสว่างจากด้านหลังทำให้เห็นเลนส์ตาและแคปซูลด้านหน้าได้ชัดเจน หลังผ่าตัด 3 สัปดาห์ ค่าสายตาที่แก้ไขแล้วดีขึ้นเป็น 20/200 และ 20/1000 ตามลำดับ4)
โรคต้อหินส่งผลโดยตรงต่อพยากรณ์การมองเห็น จึงต้องรักษาอย่างจริงจัง8)
หลังจากเกิดโรคต้อหิน ให้จัดการตามอัลกอริทึม 5 ขั้นตอนดังนี้
การรักษาด้วยยา: ยากลุ่ม beta-blockers, sympathomimetics, และ prostaglandin (PG) analogs มีประสิทธิภาพ การใช้ brimonidine (alpha-adrenergic agonist) ในทารกมีความเสี่ยงต่อการกดประสาทส่วนกลาง จึงห้ามใช้ในเด็กอายุต่ำกว่า 2 ปี หากมีความเสี่ยงต่อการบาดเจ็บที่กระจกตา ควรใช้ยาที่ไม่มีสารกันเสีย
การผ่าตัดสร้างทางระบายน้ำออกใหม่ (goniotomy/trabeculotomy): แนะนำเป็นการผ่าตัดครั้งแรก 16) มีรายงานการทำ goniotomy เพื่อป้องกัน แต่ในกรณีที่ม่านตาที่เหลือปกคลุม trabecular meshwork อาจไม่ได้ผล
การผ่าตัดกรองน้ำ (trabeculectomy): มีรายงานเพียงไม่กี่รายและระยะสั้นถึงปานกลาง ในเด็กมักได้ผลไม่ดี ภาวะ hypotony หลังผ่าตัดเกิดขึ้นประมาณ 25% 13) และมีรายงาน glaucoma ร้ายแรงหลังผ่าตัด
การผ่าตัดใส่ท่อระบายน้ำ (tube shunt surgery): สามารถใช้อุปกรณ์ชนิด Baerveldt และ Ahmed ได้ แนะนำให้ใส่ท่อในตาแบบ tangential แทนการชี้ไปที่ศูนย์กลางกระจกตาในตาที่ยังมีเลนส์ธรรมชาติ คาดว่าสามารถควบคุมความดันตาได้ดี
การจี้ทำลาย ciliary body (cyclophotocoagulation): เป็นทางเลือกสุดท้าย มีรายงานว่าการจี้เยือกแข็ง ciliary body มักทำให้เกิด hypotony เนื่องจาก ciliary body มีการพัฒนาน้อยกว่าปกติ ความเสี่ยงต่อ hypotony จึงสูงกว่าตาปกติ
เนื่องจากมีความผิดปกติของมุมลูกตาที่เป็นพื้นฐาน จึงจำเป็นต้องใช้แนวทางที่แตกต่างจากโรคต้อหินมุมเปิดทั่วไป ครั้งแรกควรเลือกการผ่าตัดสร้างทางระบายน้ำออกใหม่ ตามด้วยการผ่าตัดใส่ท่อระบายน้ำซึ่งเป็นทางเลือกที่ดี บริมอนิดีนห้ามใช้ในเด็กอายุต่ำกว่า 2 ปี และการใช้ยาต้านเมแทบอไลต์อาจทำให้ AAK แย่ลง จึงต้องพิจารณาอย่างรอบคอบ8)
ควรเริ่มการดูแลผู้มีสายตาเลือนรางตั้งแต่ระยะแรก8)
การแก้ไขค่าสายตาเป็นพื้นฐาน โดยอัตราการเกิดสายตาสั้นร่วมด้วยสูงถึง 64% ขึ้นไป
การรักษาอาการกลัวแสงมีความสำคัญต่อการพัฒนาการมองเห็นและคุณภาพชีวิต8)
ผู้ป่วยส่วนใหญ่สามารถเรียนในโรงเรียนปกติได้ แต่จำเป็นต้องมีสิ่งสนับสนุน เช่น หนังสือเรียนขยายใหญ่ อุปกรณ์แท็บเล็ต และที่วางหนังสือ การเข้าเรียนในชั้นเรียนสำหรับผู้บกพร่องทางการเห็น หรือการใช้บริการให้คำปรึกษาด้านการเลี้ยงดูบุตรและการศึกษาจากโรงเรียนคนตาบอดหรือโรงเรียนสนับสนุนพิเศษทางสายตาก็เป็นทางเลือกหนึ่ง
ตั้งแต่เดือนเมษายน พ.ศ. 2560 โรคนี้ได้รับการขึ้นทะเบียนเป็นโรคหายากที่กำหนด ดังนั้นแม้จะยังไม่ได้รับบัตรประจำตัวคนพิการทางร่างกาย หากมีความรุนแรงระดับ III ขึ้นไป ก็จะมีสิทธิ์ได้รับการช่วยเหลือค่ารักษาพยาบาลและการจัดหาอุปกรณ์ช่วยเหลือ7) อุปกรณ์ช่วยเหลือที่ครอบคลุม ได้แก่ แว่นสายตา แว่นกันแสง คอนแทคเลนส์ (รวมถึงชนิดที่มีม่านตาเทียม) แว่นขยายสำหรับผู้บกพร่องทางการเห็น ไม้เท้าปลอดภัยสำหรับผู้พิการทางสายตา และตาเทียม
PAX6 ครอบคลุมจีโนม DNA ขนาด 22 kb ซึ่งประกอบด้วย 14 เอ็กซอน และเข้ารหัสกรดอะมิโน 422 ตัว1) มีโดเมนจับกับ DNA สองโดเมน (paired domain และ paired-type homeodomain) และโดเมน PST (ที่อุดมด้วยโพรลีน ซีรีน และทรีโอนีน) ที่ปลาย C ทำหน้าที่เป็นตัวกระตุ้นการถอดรหัส
PAX6 ควบคุมการเพิ่มจำนวน การแยกตัว การเคลื่อนที่ และการยึดเกาะของเซลล์ โดยเป้าหมายของมันรวมถึงยีนที่เข้ารหัสคริสตัลลินของเลนส์และเคราตินของกระจกตา นอกเหนือจาก PAX6 เอง การแสดงออกยังคงดำเนินต่อไปในจอประสาทตา เลนส์ และกระจกตาของผู้ใหญ่ ยีน PAX6 เป็นหนึ่งในยีนควบคุมหลักที่ควบคุมการสร้างอวัยวะในระยะตัวอ่อน
การกลายพันธุ์ของ PAX6 ส่วนใหญ่ทำให้เกิด haploinsufficiency ผ่านการสลาย mRNA ที่ขึ้นกับการกลายพันธุ์ nonsense (NMD)1) การกลายพันธุ์ที่ทำให้เกิดรหัสหยุดก่อนกำหนด (PTC) เช่น การกลายพันธุ์ nonsense, frameshift และการกลายพันธุ์ splice ส่วนใหญ่ ส่งผลให้เกิดฟีโนไทป์ของ aniridia ทั่วไป
ในทางกลับกัน หาก PTC อยู่ในเอ็กซอนสุดท้ายหรือภายใน 50 คู่เบสสุดท้ายของเอ็กซอนรองสุดท้าย อาจหลบเลี่ยง NMD และโปรตีนที่ถูกตัดทอนจะถูกแปล ส่งผลให้เกิดฟีโนไทป์รุนแรงได้1)
มีรายงานกรณีที่พบได้ยากซึ่งผู้ป่วยรายเดียวกันมีการกลายพันธุ์ nonsense ของ PAX6 c.282C>A (p.Cys94*) ร่วมกับ trisomy 21 การกลายพันธุ์ PAX6 เกิดขึ้นแบบ de novo และทำให้เกิดภาวะ aniridia ทั้งสองข้างแต่กำเนิด ต้อหินแต่กำเนิด AAK และ foveal hypoplasia2)
แม้ว่าจะยังไม่มีการกำหนดความสัมพันธ์ระหว่างจีโนไทป์และฟีโนไทป์ที่ชัดเจน แต่ก็ทราบแนวโน้มบางประการ1)
จากการตรวจ gonioscopy ซีรีส์ของ Grant และ Walton พบว่าในระยะแรก iris stroma จะยื่นไปข้างหน้าบน trabecular meshwork เกิดการยึดเกาะคล้าย synechia แล้วค่อยๆ กลายเป็นแผ่นปิดจนกระทั่งมุมตาปิดสนิทในที่สุด 14) กลไกนี้เป็นสาเหตุหลักของการเกิดต้อหิน ทางพยาธิวิทยาพบว่ามีการขาดหายของกล้ามเนื้อเรียบที่เหลือ iris root และความบกพร่องของพัฒนาการของมุมตาเป็นพื้นฐาน
AAK เกิดจากภาวะพร่องเซลล์ต้นกำเนิดลิมบัส (LSCD) เป็นหลัก แต่ยังเกี่ยวข้องกับการเจริญผิดปกติของเยื่อบุกระจกตา การยึดเกาะที่ผิดปกติ การแทรกซึมของเซลล์เยื่อบุตา และการผลิตน้ำตาที่ไม่เพียงพอ การขาดเอนไซม์เมทริกซ์เมทัลโลโปรตีเนส 9 (MMP-9) ซึ่งควบคุมโดย PAX6 ทำให้เกิดการสะสมของไฟบรินและการแทรกซึมของเซลล์อักเสบ ส่งผลให้การเรียงตัวของคอลลาเจนในชั้นสโตรมาเสียไปจนสูญเสียความโปร่งใส
AAK แบ่งออกเป็น 5 ระยะ ระยะที่ 1 มีความผิดปกติเฉพาะเยื่อบุส่วนรอบนอก ระยะที่ 2 มีการเปลี่ยนแปลงของเยื่อบุแบบเข้าสู่ศูนย์กลาง (ยังไม่ถึงส่วนกลาง) ระยะที่ 3 มีการเปลี่ยนแปลงของเยื่อบุกระจกตาส่วนกลางร่วมกับเส้นเลือดงอกใหม่ที่ผิวชั้นตื้นบริเวณรอบนอก ระยะที่ 4 มีเส้นเลือดงอกใหม่ที่ผิวชั้นตื้นทั่วทั้งกระจกตา ระยะที่ 5 มีความผิดปกติของเยื่อบุทั่วทั้งกระจกตาและแผลเป็นในชั้นสโตรมาส่วนลึก10)
สถานะการกลายพันธุ์ของ PAX6 มีความสัมพันธ์กับการดำเนินโรคของ AAK ผู้ป่วยที่มีการกลายพันธุ์แบบ PTC หรือส่วนปลาย C ต่อยาว จะมี AAK ที่ดำเนินไปตามอายุ ในขณะที่การกลายพันธุ์แบบอื่นอาจทำให้เกิดโรคกระจกตาที่ไม่ดำเนินต่อ11)
กลุ่มอาการกิลเลสปีเกิดจากการกลายพันธุ์ของยีน ITPR1 3) ITPR1 เป็นสมาชิกของตระกูลตัวรับ IP3 ซึ่งสร้างช่องปล่อย Ca²⁺ และอยู่ที่เอนโดพลาสมิกเรติคูลัม การกลายพันธุ์แบบเด่นเชิงลบส่งผลต่อการสร้างและคงสภาพของกล้ามเนื้อหูรูดม่านตา ทำให้เกิดภาวะม่านตาผิดปกติเฉพาะบริเวณรอบรูม่านตาและม่านตาขยายคงที่
การทบทวนวรรณกรรมกลุ่มอาการกิลเลสปีโดย Ciaccio และคณะ (2024) จากการวิเคราะห์ผู้ป่วย 33 รายที่ได้รับการยืนยันระดับโมเลกุล พบว่าพัฒนาการด้านการเคลื่อนไหวล่าช้าแต่ดีขึ้นตามเวลา ภาวะบกพร่องทางสติปัญญาไม่พบในทุกราย โดย 17% มีสติปัญญาปกติ และอาการทางระบบประสาทไม่มีการดำเนินโรค 3)
ด้วยความแพร่หลายของเทคโนโลยีการวิเคราะห์เอ็กโซมทั้งหมด การระบุการกลายพันธุ์ของ PAX6 ใหม่ยังคงดำเนินต่อไป ณ ปี 2018 ฐานข้อมูล Human PAX6 Mutation Database มีการกลายพันธุ์ที่ลงทะเบียนไว้ 491 รายการ และตั้งแต่นั้นมามีรายงานการกลายพันธุ์ใหม่ประมาณ 250 รายการ1) กรณีที่การกลายพันธุ์ในบริเวณที่ไม่เข้ารหัสเป็นสาเหตุของภาวะไม่มีม่านตากำลังถูกระบุมากขึ้น ซึ่งคาดว่าจะช่วยอธิบายกรณีที่ไม่สามารถวินิจฉัยได้ด้วยการตรวจแบบเดิม9)
ในการผ่าตัดต้อกระจกในผู้ป่วยที่มี AAK รุนแรง เทคนิคการมองเห็นด้วยแสงย้อนจากโคมระย้าช่วยให้มองเห็นได้ดีขึ้น4) เทคนิคนี้ทำให้สามารถทำการสลายต้อกระจกด้วยคลื่นเสียงความถี่สูงได้อย่างปลอดภัยในผู้ป่วยที่มี AAK ระดับ 3–4 และช่วยให้การมองเห็นหลังผ่าตัดดีขึ้น
ประเภทของการกลายพันธุ์ของ PAX6 กำลังแสดงให้เห็นว่ารูปแบบการดำเนินของ AAK แตกต่างกัน ด้วยต้นทุนการตรวจทางพันธุกรรมที่ลดลง การทำนายแนวทางทางคลินิกและการแทรกแซงตั้งแต่เนิ่นๆ ตามชนิดของการกลายพันธุ์กำลังกลายเป็นทางเลือกที่สมจริงมากขึ้น
ในกรณีที่มีภาวะ aniridia ร่วมกับ trisomy 21 มีรายงานผู้ป่วยที่แสดงอาการค่อนข้างไม่รุนแรงแม้จะมีโรคทั้งสองร่วมกัน2) การทำความเข้าใจผลกระทบต่อฟีโนไทป์เมื่อมีความผิดปกติทางพันธุกรรมหลายอย่างในผู้ป่วยรายเดียวกันอาจนำไปสู่ข้อมูลสำคัญสำหรับการแพทย์เฉพาะบุคคลในอนาคต
การใช้ยาอ่านผ่านรหัสพันธุกรรม (ataluren) สำหรับการกลายพันธุ์ชนิด PTC ในการรักษาโรคไม่มีม่านตา (aniridia) กำลังถูกศึกษาในระดับพื้นฐานการวิจัย8) สำหรับการบำบัดด้วยยีน PAX6 การวิจัยพื้นฐานเกี่ยวกับการเติมยีนโดยใช้เวกเตอร์ AAV-PAX6 ในหนูแบบจำลองการกลายพันธุ์ Sey กำลังดำเนินอยู่ คาดว่าจะมีการพัฒนาไปสู่การทดลองทางคลินิกในอนาคต
การปลูกถ่ายแผ่นเซลล์เยื่อบุกระจกตาที่ได้จากเซลล์ iPS กำลังดำเนินการทดลองทางคลินิกทั้งในและต่างประเทศ และได้รับความสนใจในฐานะวิธีการรักษาใหม่สำหรับ AAK 8) สำหรับม่านตาเทียม (เช่น CustomFlex Artificial Iris) มีการสะสมประสบการณ์การใช้งานในต่างประเทศ คอนแทคเลนส์ที่มีม่านตาเทียมเป็นอุปกรณ์เสริมอยู่ภายใต้การครอบคลุมของประกันสุขภาพ
การทำความเข้าใจสถานการณ์จริงผ่านการสะสมข้อมูลทะเบียนขนาดใหญ่ในประเทศและการปรับปรุงคุณภาพของหลักฐานเป็นประเด็นสำคัญในอนาคต 8) คาดว่าจะสามารถทำนายการดำเนินโรคของ AAK ตามการกลายพันธุ์ทางพันธุกรรมแต่ละรายและการปรับการแทรกแซงในระยะเริ่มต้นให้เหมาะสม