Aniridia isolata
Frequenza: circa 2/3 del totale.
Modalità di trasmissione: autosomica dominante (AD).
Caratteristiche: causata da mutazioni del gene PAX6. Non associata a sintomi sistemici. Penetranza completa ma espressività variabile.

L’aniridia è una rara malattia congenita caratterizzata da ipoplasia o assenza parziale dell’iride. Il termine “aniridia” è un nome improprio, poiché all’esame gonioscopico o con ecografia biomicroscopica (UBM) si osservano quasi sempre frammenti di tessuto irideo.
La prevalenza è di circa 1/40.000-1/100.000, senza differenze razziali o di genere significative 1). È classificata come Q13.1 nell’ICD-10.
Si tratta di una malattia panoculare che coinvolge non solo l’iride, ma anche cornea, cristallino, angolo camerulare, fovea e nervo ottico 1), presentando diverse complicanze oculari che minacciano la vista. La prognosi visiva è generalmente sfavorevole, con un’acuità visiva corretta spesso intorno a 0,1. Il riflesso pupillare è assente, ma la reazione accomodativa è preservata; nel 60-90% dei casi è bilaterale.
Si riconoscono i seguenti tre fenotipi.
Aniridia isolata
Frequenza: circa 2/3 del totale.
Modalità di trasmissione: autosomica dominante (AD).
Caratteristiche: causata da mutazioni del gene PAX6. Non associata a sintomi sistemici. Penetranza completa ma espressività variabile.
Sindrome WAGR
Frequenza: una parte dei casi sporadici.
Modalità di trasmissione: delezione contigua di PAX6 e WT1.
Caratteristiche: associata a tumore di Wilms, anomalie genitourinarie e ritardo mentale. Rischio tumorale fino al 50%.
Sindrome di Gillespie
Frequenza: circa il 2% del totale.
Modalità di trasmissione: mutazioni del gene ITPR1.
Caratteristiche: associata a atassia cerebellare e disabilità intellettiva. Caratteristica anomalia iridea con midriasi fissa 3).
L’aniridia sporadica rappresenta circa 1/3 del totale ed è causata da delezioni de novo in 11p13 che includono PAX6. Se la delezione si estende al gene WT1 adiacente, causa la sindrome WAGR 1). Il 25-30% dei casi di aniridia sporadica sviluppa tumore di Wilms, con un rischio relativo riportato di 67.
PAX6 è un gene master control della formazione dell’occhio, coinvolto nello sviluppo dell’occhio, del tubo neurale, del bulbo olfattivo, delle isole di Langerhans pancreatiche e dell’epitelio olfattivo. La malattia si manifesta con aploinsufficienza (perdita di funzione di un allele); anomalie bialleliche sono letali in epoca embrionale. Dal 2017 è stata designata come malattia rara ai sensi della legge sulle malattie difficili, e per i gradi di gravità III o superiore (vedere la sezione Diagnosi ed esami per i dettagli) è ammissibile per il sussidio delle spese mediche 7).
I casi sporadici (nuove mutazioni) rappresentano circa 1/3 del totale e possono verificarsi anche senza storia familiare. Nei casi sporadici esiste la possibilità di sindrome WAGR, pertanto sono importanti il test genetico e lo screening del tumore di Wilms tramite ecografia addominale.
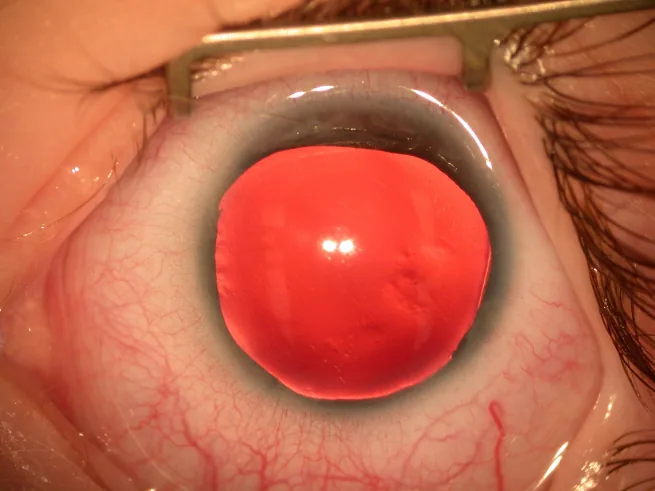
La maggior parte dei casi di aniridia viene scoperta alla nascita per anomalie dell’iride e della pupilla, o durante l’infanzia per nistagmo.
Il fenotipo varia tra famiglie e all’interno della stessa famiglia, ma le differenze tra occhio destro e sinistro sono generalmente minime.
A causa principalmente dell’ipoplasia foveale, l’acuità visiva corretta è spesso compresa tra 0,1 e 0,2. La prognosi visiva è particolarmente sfavorevole in caso di ipoplasia maculare associata. La correzione refrattiva e la riabilitazione visiva fin dalla prima infanzia sono importanti per lo sviluppo visivo.
PAX6 è espresso non solo nei tessuti oculari, ma anche nel sistema nervoso centrale, nelle isole di Langerhans del pancreas e nell’epitelio olfattivo; pertanto, possono essere presenti le seguenti complicanze extraoculari 8).
I fattori importanti che determinano la funzione visiva sono glaucoma, ipoplasia maculare, nistagmo, cheratopatia, cataratta e anomalie dell’iride. Poiché il danno del campo visivo e dell’acuità visiva causato dal glaucoma è irreversibile, la gestione della pressione intraoculare è fondamentale durante il follow-up.8)
La maggior parte dei casi di aniridia congenita è causata da mutazioni eterozigoti del gene PAX6, situato sul braccio corto del cromosoma 11 (11p13). L’aploinsufficienza di PAX6 è il principale meccanismo patogenetico.1)
Il gene PAX6 è un gene maestro per la formazione dell’occhio e svolge un ruolo importante nello sviluppo dell’occhio, del tubo neurale, del bulbo olfattivo e del pancreas. Per il normale sviluppo dell’occhio sono necessarie due copie di PAX6; la perdita di funzione di una sola copia è sufficiente per causare l’aniridia.1)
In uno studio di coorte su pazienti cinesi, nel 96,9% dei casi è stata identificata una mutazione causale nel gene PAX6.1) Nell’aniridia tipica, nel 96% dei casi vengono rilevate mutazioni che inducono la degradazione dell’mRNA mediata da mutazioni nonsenso (NMD) o delezioni su larga scala.1)
Dal punto di vista patologico, la muscolatura liscia è assente tranne che alla radice dell’iride, e si osserva uno sviluppo anomalo dell’angolo camerulare. Si riscontra una disfunzione delle cellule staminali epiteliali corneali, con anomalie dell’epitelio e della membrana di Bowman, e formazione di panno vascolarizzato.
Di seguito è riportata la distribuzione delle mutazioni di PAX6 che causano il fenotipo dell’aniridia.
| Tipo di mutazione | Frequenza |
|---|---|
| Mutazione nonsenso | Circa 39% |
| Mutazione frameshift | Circa 25% |
| Mutazione di splicing | Circa 13% |
| Mutazione missenso | Circa il 12% |
Le mutazioni read-through (mutazioni di estensione C-terminale) rappresentano circa il 5% e producono una proteina PAX6 anormalmente allungata a causa della conversione del codone di stop in un codone di traduzione 6). Le mutazioni di estensione C-terminale sono spesso associate a grave ipoplasia dell’iride e grave compromissione visiva 1)6).
Le mutazioni genetiche sono per lo più di tipo PTC, ma sono state riportate anche mutazioni missenso 7). Per quanto riguarda l’utilità dei test genetici, il sequenziamento Sanger o NGS rileva mutazioni in quasi l’85% dei casi di aniridia isolata. Inoltre, MLPA o CMA rilevano delezioni all’interno del gene PAX6 o della regione cis-regolatoria in quasi il 15% dei casi 8).
Wang (2023) ha identificato una nuova mutazione frameshift c.640_646del (p.R214Pfs*28) e ha riportato un caso con assenza completa dell’iride, ipoplasia foveale, ectopia lentis e distacco di retina 1).
Ratna et al. (2022) hanno identificato una mutazione read-through c.1268A>T (p.*423L) in una famiglia indiana. I soggetti affetti presentavano aniridia completa, nistagmo, ipoplasia foveale, AAK, sublussazione superiore del cristallino, miopia elevata e atrofia ottica, mostrando un fenotipo grave dovuto alla mutazione di estensione C-terminale 6).
Nell’aniridia sporadica, le grandi delezioni che includono il gene WT1 oltre a PAX6 causano la sindrome WAGR. Il rischio di tumore di Wilms in presenza di delezione di WT1 arriva fino al 50% 1). Se si sospetta la sindrome WAGR, i test genetici possono confermare le delezioni di PAX6 e WT1, consentendo la valutazione del rischio di tumore di Wilms e il monitoraggio del ritardo dello sviluppo 8). La valutazione della regione WT1 mediante test genetici è essenziale; si stima che il 30% dei casi sporadici sviluppi il tumore di Wilms entro i 5 anni di età. Poiché il gene WT1 si trova vicino a PAX6, la delezione del braccio corto del cromosoma 11 (delezione 11p13) che coinvolge entrambi i geni provoca aniridia associata a tumore di Wilms.
La sindrome di Gillespie è causata da mutazioni eterozigoti dominanti negative o bialleliche del gene ITPR1 3). Finora sono stati riportati 37 casi con diagnosi molecolare confermata, e il residuo Gly2554 è noto come hot spot 3).
Secondo i criteri diagnostici per l’aniridia (2020), la diagnosi viene confermata in base ai seguenti criteri7).
A. Sintomi
B. Reperti strumentali
C. Diagnosi differenziale
E. Test genetici: mutazione patogenetica del gene PAX6 o delezione della regione 11p13
Categorie diagnostiche7):
La classificazione della gravità per il riconoscimento di malattia rara è definita in 4 stadi7).
| Gravità | Definizione |
|---|---|
| Grado I | Coinvolgimento di un occhio, l’altro sano |
| Grado II | Coinvolgimento bilaterale, migliore acuità visiva corretta ≥ 0.3 |
| Grado III | Coinvolgimento bilaterale, migliore acuità visiva corretta ≥ 0.1 e < 0.3 |
| Grado IV | Coinvolgimento bilaterale, migliore acuità visiva corretta < 0.1 |
Anche per i gradi I-III, se è presente restringimento del campo visivo dovuto a glaucoma (campo visivo centrale residuo entro 20 gradi con lo stimolo Goldmann I/4), si passa al grado di gravità superiore. Il grado III o superiore è idoneo per il sussidio medico 7).
La diagnosi clinica è semplice se si osserva un difetto o un’ipoplasia dell’iride alla lampada a fessura. La valutazione del tessuto irideo residuo viene eseguita con gonioscopia o microscopia ultrasonica biomicroscopica. Si verifica anche la presenza di anomalie dello sviluppo dell’angolo della camera anteriore.
Valutare sistematicamente le seguenti complicanze oculari.
L’obiettivo più importante nella valutazione genetica dell’aniridia è verificare se la delezione di PAX6 si estende al gene WT11). Il sequenziamento dell’intero esoma e la tecnica MLPA valutano mutazioni e delezioni nelle regioni PAX6 e WT11)2).
Nell’aniridia sporadica, la valutazione del rischio di tumore di Wilms dovuto a delezioni del gene WT1 è direttamente correlata alla prognosi vitale1). Anche nei casi familiari, a causa della variabilità fenotipica, si raccomanda la diagnosi definitiva tramite test genetico e consulenza genetica.
Non esiste una terapia curativa per l’aniridia. La gestione si basa sulla riabilitazione visiva per massimizzare la vista residua e sul trattamento individuale delle complicanze8).
Il trapianto di cornea per l’opacità stromale deve essere valutato con cautela8).
Il trapianto di cornea può migliorare a breve termine l’acuità visiva, ma il miglioramento è limitato a causa di comorbidità come l’ipoplasia maculare. A lungo termine, la prognosi visiva è sfavorevole a causa della progressione del glaucoma e del fallimento del trapianto.
Nella carenza di cellule staminali epiteliali corneali, si considera il trattamento chirurgico 8).
La chirurgia della cataratta viene presa in considerazione in base al grado di opacità e fotofobia 8).
La cataratta si sviluppa nel 50-85% dei casi entro i 20 anni di età. L’intervento viene pianificato in base all’intensità dell’opacità e della fotofobia. È stato riportato un miglioramento dell’acuità visiva nel 66-100% dei casi operati, ma è necessario prestare attenzione ai seguenti punti.
A causa della fragilità della zonula di Zinn, l’impianto di lente intraoculare richiede un’attenta valutazione.
Hu et al. (2024) hanno eseguito la facoemulsificazione con illuminazione retrograda a candelabro in due casi di aniridia congenita con grave AAK. Nonostante la difficoltà di visualizzazione intraoperatoria a causa dell’opacità corneale, l’illuminazione posteriore ha permesso una chiara visualizzazione del cristallino e della capsula anteriore, con miglioramento dell’acuità visiva corretta a 20/200 e 20/1000 rispettivamente a 3 settimane dall’intervento 4).
Il glaucoma influisce direttamente sulla prognosi visiva, pertanto va trattato attivamente 8).
Dopo l’insorgenza del glaucoma, la gestione si basa sul seguente algoritmo a 5 fasi.
Terapia farmacologica: beta-bloccanti, simpaticomimetici e prostaglandine (PG) sono efficaci. La brimonidina (agonista alfa-adrenergico) nei neonati è controindicata sotto i 2 anni per il rischio di depressione del sistema nervoso centrale. In caso di sospetta tossicità epiteliale corneale, utilizzare preparati senza conservanti.
Chirurgia di ricostruzione del deflusso (goniotomia/trabeculotomia): raccomandata come primo intervento 16). Esistono anche segnalazioni di goniotomia profilattica. Tuttavia, può essere inefficace nei casi in cui l’iride residua copre il trabecolato.
Chirurgia filtrante (trabeculectomia): limitata a segnalazioni su piccoli campioni e medio-breve termine. Nei bambini, i risultati tendono a essere scadenti, con una incidenza di ipotonia postoperatoria fino al 25% 13). Sono state segnalate anche glaucoma maligno postoperatorio.
Impianto di dispositivo per glaucoma (chirurgia di shunt tubulare): dispositivi tipo Baerveldt e Ahmed sono disponibili. Negli occhi facchici, si raccomanda l’inserimento del tubo in direzione tangenziale anziché verso il centro corneale. Ci si può aspettare un buon controllo della pressione intraoculare.
Ciclocoagulazione: ultima risorsa. La ciclocriocoagulazione ha portato a ipotonia in molti casi. A causa dell’ipoplasia del corpo ciliare, il rischio di ipotonia è maggiore rispetto agli occhi sani.
A causa dell’anomalia dello sviluppo dell’angolo, è necessario un approccio diverso rispetto al glaucoma ad angolo aperto comune. Come primo intervento si sceglie la ricostruzione della via di deflusso, seguita dallo shunt con tubo come buona opzione. La brimonidina è controindicata nei bambini di età inferiore a 2 anni e l’uso di antimetaboliti può peggiorare l’AAK, pertanto è necessaria una valutazione attenta 8).
La cura della bassa visione va introdotta precocemente 8).
La correzione refrattiva è fondamentale; il tasso di complicanze della miopia è superiore al 64%.
Il trattamento della fotofobia è importante per preservare lo sviluppo della funzione visiva e la qualità della vita 8).
La maggior parte dei pazienti può frequentare classi normali, ma necessita di supporti come libri di testo ingranditi, tablet e leggii. Sono opzioni anche la frequenza di classi per ipovedenti o la consulenza educativa e per l’infanzia presso scuole per non vedenti o scuole speciali per la vista.
Dal aprile 2017, questa condizione è stata riconosciuta come malattia rara designata, quindi anche senza il certificato di disabilità, i pazienti con gravità di grado III o superiore possono ricevere assistenza medica e sussidi per ausili 7). Gli ausili coperti includono occhiali correttivi, occhiali schermanti, lenti a contatto (incluse quelle con iride artificiale), occhiali per ipovedenti, bastoni di sicurezza per non vedenti e protesi oculari.
PAX6 si estende su 22 kb di DNA genomico contenente 14 esoni e codifica 422 amminoacidi 1). Possiede due domini di legame al DNA (dominio paired e homeodominio paired) e un dominio PST (ricco di prolina, serina e treonina) all’estremità C-terminale che funge da attivatore trascrizionale.
PAX6 regola la proliferazione, differenziazione, migrazione e adesione cellulare, e i suoi bersagli includono lo stesso PAX6, oltre ai geni che codificano per le cristalline del cristallino e le cheratine corneali. L’espressione continua anche nella retina, nel cristallino e nella cornea dell’adulto. PAX6 è uno dei geni master control che governano la differenziazione degli organi durante la vita embrionale.
La maggior parte delle mutazioni di PAX6 causa aploinsufficienza attraverso la degradazione dell’mRNA mediata da mutazioni nonsenso (NMD) 1). Le mutazioni che introducono un codone di stop prematuro (PTC) (mutazioni nonsenso, frameshift e la maggior parte delle mutazioni di splicing) portano al tipico fenotipo dell’aniridia.
D’altra parte, se il PTC si trova nell’esone finale o entro 50 bp dall’estremità del penultimo esone, può sfuggire all’NMD e tradurre una proteina troncata, portando potenzialmente a un fenotipo grave 1).
È stato riportato un raro caso di mutazione nonsenso di PAX6 c.282C>A (p.Cys94*) associata a trisomia 21 nello stesso paziente. La mutazione PAX6 è insorta de novo, causando aniridia bilaterale completa, glaucoma congenito, AAK e ipoplasia foveale 2).
Sebbene non sia stata stabilita una chiara correlazione genotipo-fenotipo, sono note alcune tendenze 1).
Nella serie di gonioscopie di Grant e Walton, è stato dimostrato che inizialmente lo stroma dell’iride si estende anteriormente sul trabecolato formando aderenze simili a sinechie, che gradualmente diventano a lamina e portano infine all’occlusione dell’angolo14). Questo meccanismo è il principale fattore nello sviluppo del glaucoma. Patologicamente, la base è costituita da un difetto della muscolatura liscia con risparmio della radice dell’iride e da uno sviluppo incompleto dell’angolo.
L’AAK è causata principalmente da una deficienza di cellule staminali del limbo (LSCD), ma sono coinvolti anche una differenziazione anomala dell’epitelio corneale, difetti di adesione, infiltrazione di cellule congiuntivali e produzione insufficiente di lacrime. La carenza di metalloproteinasi-9 della matrice (MMP-9), regolata da PAX6, provoca accumulo di fibrina e infiltrazione di cellule infiammatorie, portando a una perdita di trasparenza dovuta a un’alterazione dell’architettura del collagene stromale.
L’AAK è classificata in 5 stadi. Lo stadio I presenta anomalie solo dell’epitelio periferico, lo stadio II mostra cambiamenti epiteliali centripeti (senza raggiungere il centro), lo stadio III presenta alterazioni epiteliali corneali centrali e neovascolarizzazione superficiale periferica, lo stadio IV mostra neovascolarizzazione superficiale di tutta la cornea e lo stadio V presenta anomalie epiteliali corneali totali e cicatrici stromali profonde10).
Esiste una correlazione tra lo stato della mutazione di PAX6 e la progressione dell’AAK. Nei pazienti con mutazioni PTC o estensione del C-terminale, l’AAK progredisce in modo dipendente dall’età, mentre in altri tipi di mutazione può presentarsi come cheratopatia non progressiva11).
La sindrome di Gillespie è causata da mutazioni nel gene ITPR13). ITPR1 è un membro della famiglia dei recettori IP3, forma canali di rilascio del Ca²⁺ ed è localizzato nel reticolo endoplasmatico. Le mutazioni dominanti negative influenzano la formazione e il mantenimento del muscolo sfintere dell’iride, portando a un’ipoplasia dell’iride specifica attorno alla pupilla e a midriasi fissa.
Nella revisione della letteratura sulla sindrome di Gillespie di Ciaccio et al. (2024), l’analisi di 33 casi confermati molecolarmente ha mostrato che lo sviluppo motorio è ritardato ma migliora nel tempo, il deficit intellettivo non è presente in tutti i casi e il 17% ha un’intelligenza normale, e i segni neurologici sono non progressivi3).
Con la diffusione della tecnologia di sequenziamento dell’intero esoma, continuano a essere identificate nuove mutazioni di PAX6. Nel 2018, il database Human PAX6 Mutation Database conteneva 491 mutazioni e da allora sono state riportate circa 250 nuove mutazioni1). Stanno emergendo anche casi in cui mutazioni in regioni non codificanti causano aniridia, aprendo la strada alla diagnosi di casi precedentemente non diagnosticabili con i test tradizionali9).
Nei casi di cataratta con AAK grave, la tecnica di visualizzazione con illuminazione a candelabro inversa è utile4). Questa procedura consente una facoemulsificazione sicura anche in pazienti con AAK di grado 3-4, portando a un miglioramento della vista post-operatoria.
Sta diventando chiaro che diversi tipi di mutazioni di PAX6 portano a diversi modelli di progressione dell’AAK. Con la riduzione dei costi dei test genetici, la previsione del decorso clinico basata sul tipo di mutazione e l’intervento precoce stanno diventando opzioni realistiche.
In casi di aniridia e trisomia 21 concomitanti, nonostante la coesistenza di entrambe le malattie, sono stati riportati esempi con un decorso relativamente lieve2). Comprendere l’impatto sul fenotipo quando più disturbi genetici coesistono nello stesso paziente potrebbe fornire importanti spunti per la medicina personalizzata futura.
L’applicazione del farmaco read-through (ataluren) per mutazioni di tipo PTC nell’aniridia è in fase di studio a livello di ricerca di base8). Per quanto riguarda la terapia genica di PAX6, sono in corso studi di base sul ripristino genico tramite vettori AAV-PAX6 in modelli murini con mutazione Sey. Ci si aspetta un futuro sviluppo verso studi clinici.
Studi clinici sul trapianto di foglietti di cellule epiteliali corneali derivati da iPS sono in corso sia in Giappone che all’estero, attirando l’attenzione come nuovo trattamento per l’AAK8). Per quanto riguarda l’iride artificiale (come CustomFlex Artificial Iris), l’esperienza d’uso all’estero si sta accumulando. Le lenti a contatto con iride artificiale come protesi sono coperte dall’assicurazione sanitaria.
L’accumulo di dati da registri su larga scala in Giappone per comprendere la situazione reale e migliorare la qualità delle prove è una questione importante per il futuro8). Ci si aspetta un’ottimizzazione della previsione della progressione dell’AAK e dell’intervento precoce basata sulle singole mutazioni genetiche.