آنیریدیای ایزوله
فراوانی: حدود دو سوم کل موارد.
الگوی وراثت: اتوزومال غالب (AD).
ویژگیها: ناشی از جهش در ژن PAX6. بدون علائم سیستمیک. نفوذ کامل اما بیان متغیر.

آنیریدیا (Aniridia) یک بیماری مادرزادی نادر است که با درجات مختلفی از هیپوپلازی یا فقدان عنبیه مشخص میشود. نام «آنیریدیا» یک نام اشتباه است، زیرا قطعاتی از بافت عنبیه تقریباً همیشه در گونیوسکوپی یا میکروسکوپ اولتراسوند بیومیکروسکوپی (UBM) قابل مشاهده است.
شیوع آن حدود 1 در 40,000 تا 1 در 100,000 تخمین زده میشود و تفاوت نژادی یا جنسی قابل توجهی گزارش نشده است1). در ICD-10 تحت کد Q13.1 طبقهبندی میشود.
این یک بیماری پاناکولار است که نه تنها عنبیه، بلکه قرنیه، عدسی، زاویه اتاق قدامی، فووئا و عصب بینایی را نیز تحت تأثیر قرار میدهد1) و عوارض چشمی متعددی را ایجاد میکند که بینایی را تهدید میکند. پیشآگهی بینایی عموماً ضعیف است و اغلب حدت بینایی اصلاحشده حدود 0.1 است. واکنش مردمک از بین رفته است اما واکنش تطابقی حفظ میشود و 60 تا 90 درصد موارد دوطرفه هستند.
سه فنوتیپ زیر شناخته شده است.
آنیریدیای ایزوله
فراوانی: حدود دو سوم کل موارد.
الگوی وراثت: اتوزومال غالب (AD).
ویژگیها: ناشی از جهش در ژن PAX6. بدون علائم سیستمیک. نفوذ کامل اما بیان متغیر.
سندرم WAGR
فراوانی: بخشی از موارد پراکنده.
الگوی وراثت: حذف مجاور ژنهای PAX6 و WT1.
ویژگیها: همراه با تومور ویلمز، ناهنجاریهای دستگاه ادراری-تناسلی و عقبماندگی ذهنی. خطر تومور تا ۵۰٪.
سندرم گیلزپی
فراوانی: حدود ۲٪ از کل موارد.
الگوی وراثت: جهش در ژن ITPR1.
ویژگیها: همراه با آتاکسی مخچهای و ناتوانی ذهنی. ناهنجاری مشخص عنبیه با میدریاز ثابت مشخصه آن است3).
آنیریدیای پراکنده حدود یکسوم موارد را تشکیل میدهد و ناشی از حذف de novo در ناحیه 11p13 شامل PAX6 است. اگر حذف به ژن مجاور WT1 گسترش یابد، باعث سندرم WAGR میشود1). ۲۵ تا ۳۰٪ از موارد آنیریدیای پراکنده به تومور ویلمز مبتلا میشوند و خطر نسبی ۶۷ گزارش شده است.
PAX6 یک ژن کنترلکننده اصلی در تشکیل چشم است و در رشد چشم، لوله عصبی، پیاز بویایی، جزایر لانگرهانس پانکراس و اپیتلیوم بویایی نقش دارد. بیماری با از دست دادن عملکرد یک آلل (نارسایی هاپلوئید) ایجاد میشود و ناهنجاری در هر دو آلل منجر به مرگ جنینی میگردد. در سال ۲۰۱۷ به عنوان بیماری نادر تحت قانون بیماریهای نادر تعیین شد و موارد با شدت درجه III یا بالاتر (برای جزئیات به بخش تشخیص و آزمایش مراجعه کنید) مشمول کمک هزینه پزشکی میشوند7).
موارد پراکنده (جهش جدید) حدود یک سوم موارد را تشکیل میدهند و بدون سابقه خانوادگی بروز میکنند. در موارد پراکنده، احتمال سندرم WAGR وجود دارد، بنابراین آزمایش ژنتیک و غربالگری تومور ویلمز با سونوگرافی شکم اهمیت دارد.
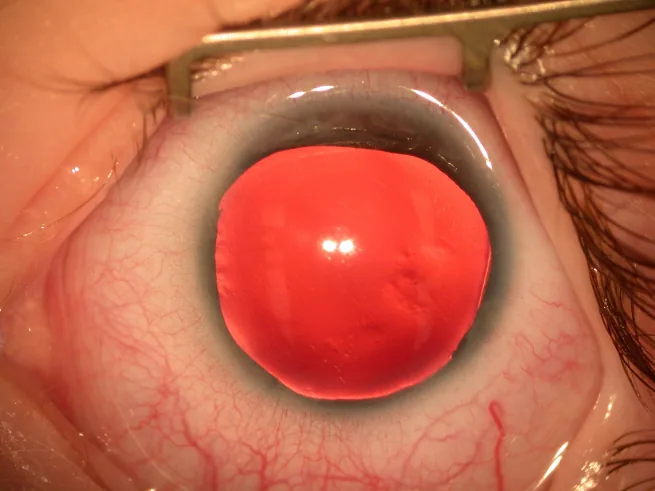
بیشتر موارد آنیریدیا با ناهنجاری عنبیه یا مردمک در بدو تولد، یا با نیستاگموس در دوران نوزادی تشخیص داده میشود.
فنوتیپ بین خانوادهها و درون یک خانواده متفاوت است، اما تفاوت بین چشمهای راست و چپ معمولاً اندک است.
نقص تشکیل فووئا علت اصلی است و حدت بینایی اصلاحشده معمولاً حدود 0.1 تا 0.2 است. در صورت همراهی با هیپوپلازی ماکولا، پیشآگهی بینایی به ویژه ضعیف است. اصلاح عیوب انکساری و مراقبتهای کمبینایی از دوران نوزادی برای رشد بینایی مهم است.
از آنجایی که PAX6 علاوه بر بافت چشم در سیستم عصبی مرکزی، جزایر لانگرهانس پانکراس و اپیتلیوم بویایی نیز بیان میشود، ممکن است عوارض خارج چشمی زیر مشاهده شود8).
عوامل مهم تعیینکننده عملکرد بینایی عبارتند از: گلوکوم، هیپوپلازی ماکولا، نیستاگموس، کراتوپاتی، آب مروارید و ناهنجاری عنبیه. از آنجایی که آسیب میدان بینایی و کاهش بینایی ناشی از گلوکوم غیرقابل برگشت است، مدیریت فشار چشم در پیگیریها از اهمیت بالایی برخوردار است8).
اکثر موارد آنیریدی مادرزادی ناشی از جهشهای هتروزیگوت در ژن PAX6 واقع در بازوی کوتاه کروموزوم ۱۱ (11p13) است. مکانیسم اصلی بیماری، هاپلوناکافی PAX6 میباشد1).
ژن PAX6 یک ژن کنترلکننده اصلی در تکامل چشم است و نقش مهمی در رشد چشم، لوله عصبی، پیاز بویایی و پانکراس ایفا میکند. برای رشد طبیعی چشم، دو کپی از PAX6 مورد نیاز است و از دست دادن عملکرد تنها یک کپی منجر به بروز آنیریدی میشود1).
در یک مطالعه کوهورت بر روی بیماران چینی، در 96.9% موارد جهشهای عامل در ژن PAX6 شناسایی شده است 1). در آنیریدیای کلاسیک، جهشهایی که تخریب وابسته به کدون بیمعنی (NMD) را القا میکنند یا حذفهای بزرگ در 96% موارد یافت میشوند 1).
از نظر آسیبشناسی، عضله صاف به جز ریشه عنبیه از دست رفته و زاویه اتاق قدامی دچار ناهنجاری رشدی است. ناهنجاری در عملکرد سلولهای بنیادی اپیتلیال قرنیه دیده میشود که منجر به ناهنجاری در اپیتلیوم و غشای بومن و تشکیل پانوس عروقی میشود.
توزیع جهشهای PAX6 که باعث فنوتیپ آنیریدیا میشوند در زیر نشان داده شده است.
| نوع جهش | فراوانی |
|---|---|
| جهش بیمعنی | حدود ۳۹٪ |
| جهش تغییر چارچوب | حدود ۲۵٪ |
| جهش پیرایش | حدود ۱۳٪ |
| جهش نادرست (میسسنس) | حدود ۱۲٪ |
جهشهای رانآن (جهشهای افزایش طول C-ترمینال) حدود ۵٪ موارد را تشکیل میدهند و در آنها کدون توقف به یک کدون ترجمهای تبدیل شده و پروتئین PAX6 به طور غیرطبیعی طویل تولید میشود6). جهشهای افزایش طول C-ترمینال اغلب با هیپوپلازی شدید عنبیه و اختلال بینایی شدید همراه هستند1)6).
جهشهای ژنتیکی عمدتاً از نوع جهشهای PTC هستند و مواردی از جهشهای نادرست (میسسنس) نیز گزارش شده است7). در مورد کارایی آزمایش ژنتیکی، با توالییابی سنگر یا NGS، نزدیک به ۸۵٪ موارد آنیریدیای ایزوله دارای جهش تشخیص داده میشوند. علاوه بر این، با MLPA یا CMA، حذفهای درون ژنی PAX6 یا ناحیه تنظیمی سیس در نزدیک به ۱۵٪ موارد شناسایی میشود8).
Wang (2023) یک جهش جدید از نوع جابجایی چارچوب به نام c.640_646del (p.R214Pfs*28) را شناسایی کرد و موردی را گزارش داد که با فقدان کامل عنبیه، هیپوپلازی فووئا، جابجایی عدسی و جداشدگی شبکیه همراه بود1).
Ratna و همکاران (2022) یک جهش run-on c.1268A>T (p.*423L) را در یک خانواده هندی شناسایی کردند. افراد مبتلا آنیریدی کامل، نیستاگموس، هیپوپلازی فووآ، AAK، سابلوکساسیون فوقانی عدسی، نزدیکبینی شدید و آتروفی عصب بینایی را نشان دادند که فنوتیپ شدید ناشی از جهش طویلسازی C-ترمینال را نشان میدهد 6).
در آنیریدی پراکنده، حذف بزرگ شامل ژن WT1 علاوه بر PAX6 باعث سندرم WAGR میشود. خطر تومور ویلمز در صورت حذف WT1 تا 50٪ است 1). در صورت مشکوک بودن به سندرم WAGR، آزمایش ژنتیکی برای تأیید حذف PAX6 و WT1 انجام میشود که امکان ارزیابی خطر تومور ویلمز و پیگیری تأخیر رشدی را فراهم میکند 8). ارزیابی ناحیه WT1 با آزمایش ژنتیکی ضروری است و 30٪ موارد پراکنده تا سن 5 سالگی به تومور ویلمز مبتلا میشوند. از آنجایی که ژن WT1 در مجاورت PAX6 قرار دارد، حذف بازوی کوتاه کروموزوم 11 (حذف 11p13) که هر دو را شامل میشود، باعث همراهی آنیریدی با تومور ویلمز میگردد.
سندرم گیلزپی در اثر جهش هتروزیگوت غالب-منفی یا جهش دو آللی در ژن ITPR1 ایجاد میشود3). تاکنون ۳۷ مورد با تشخیص مولکولی تأیید شده گزارش شده است و باقیمانده Gly2554 به عنوان یک نقطه داغ شناخته میشود3).
بر اساس معیارهای تشخیصی آنیریدیا (۲۰۲۰)، تشخیص با معیارهای زیر تأیید میشود7).
الف. علائم
B. یافتههای آزمایشگاهی
ج. بیماریهایی که باید افتراق داده شوند
E. آزمایش ژنتیکی: جهش بیماریزا در ژن PAX6 یا حذف ناحیه 11p13
ردههای تشخیصی7):
طبقهبندی شدت برای تشخیص بیماری نادر به چهار مرحله زیر تعریف شده است7).
| شدت | تعریف |
|---|---|
| درجه I | درگیری یک چشم، چشم دیگر سالم |
| درجه II | درگیری هر دو چشم، حدت بینایی اصلاحشده چشم بهتر 0.3 یا بیشتر |
| درجه III | درگیری هر دو چشم، حدت بینایی اصلاحشده چشم بهتر بین 0.1 تا کمتر از 0.3 |
| درجه IV | درگیری هر دو چشم، حدت بینایی اصلاحشده چشم بهتر کمتر از 0.1 |
حتی در درجات I تا III، در صورت وجود تنگی میدان بینایی ناشی از گلوکوم و غیره (میدان بینایی باقیمانده مرکزی 20 درجه یا کمتر با هدف Goldmann I/4)، شدت یک درجه افزایش مییابد. شدت درجه III یا بالاتر مشمول کمک هزینه پزشکی است 7).
تشخیص بالینی با مشاهده فقدان یا ناقصبودن عنبیه با استفاده از میکروسکوپ اسلیت لامپ آسان است. ارزیابی بافت باقیمانده عنبیه با گونیوسکوپی یا میکروسکوپ اولتراسوند بیومیکروسکوپی انجام میشود. همچنین وجود ناهنجاری رشدی زاویه اتاق قدامی بررسی میشود.
عوارض چشمی زیر به صورت سیستماتیک ارزیابی میشوند:
مهمترین هدف در ارزیابی ژنتیکی آنیریدیا، تأیید این است که آیا حذف PAX6 تا ژن WT1 گسترش یافته است یا خیر 1). با استفاده از توالییابی کامل اگزوم یا روش MLPA، جهشها و حذفهای ناحیه PAX6 و WT1 ارزیابی میشوند 1)2).
در آنیریدی پراکنده، ارزیابی خطر تومور ویلمز ناشی از حذف ژن WT1 مستقیماً با پیشآگهی حیاتی مرتبط است 1). حتی در موارد خانوادگی، به دلیل تنوع فنوتیپی، تشخیص قطعی با آزمایش ژنتیکی و مشاوره ژنتیک توصیه میشود.
هیچ درمان قطعی برای آنیریدیا وجود ندارد. مراقبت از دید کم برای استفاده حداکثری از بینایی باقیمانده و درمان جداگانه هر عارضه، محور اصلی مدیریت این بیماری است8).
پیوند قرنیه برای کدورت استرومای قرنیه باید با احتیاط انجام شود8).
اگرچه پیوند قرنیه ممکن است در کوتاهمدت بهبود بینایی ایجاد کند، اما به دلیل عوارض همراه مانند هیپوپلازی ماکولا، بهبود محدود است. در بلندمدت، پیشرفت گلوکوم و نارسایی پیوند منجر به پیشآگهی ضعیف بینایی میشود.
در موارد نارسایی سلولهای بنیادی اپیتلیوم قرنیه، درمان جراحی مد نظر قرار میگیرد8).
جراحی آب مروارید با توجه به درجه کدورت و حساسیت به نور بررسی میشود8).
آب مروارید در ۵۰ تا ۸۵٪ موارد تا سن ۲۰ سالگی بروز میکند. جراحی بر اساس شدت کدورت و حساسیت به نور برنامهریزی میشود. گزارش شده است که در ۶۶ تا ۱۰۰٪ موارد جراحی، بهبود بینایی مشاهده شده است، اما نکات زیر باید مورد توجه قرار گیرند:
به دلیل ضعف رباطهای زین، کاشت لنز داخل چشمی نیاز به اندیکاسیون دقیق دارد.
هو و همکاران (2024) در دو مورد آنیریدی مادرزادی همراه با AAK شدید، عمل فیکوامولسیفیکاسیون با کمک روشنایی معکوس لوستر انجام دادند. با وجود کدورت قرنیه که تجسم معمول حین عمل را دشوار میکرد، روشنایی از پشت امکان تجسم واضح عدسی و کپسول قدامی را فراهم کرد و در ۳ هفته پس از عمل، حدت بینایی اصلاحشده به ترتیب به 20/200 و 20/1000 بهبود یافت4).
گلوکوم به طور مستقیم بر پیشآگهی بینایی تأثیر میگذارد، بنابراین باید به طور فعال درمان شود 8).
پس از بروز گلوکوم، مدیریت بر اساس الگوریتم ۵ مرحلهای زیر انجام میشود.
دارودرمانی: مسدودکنندههای بتا، محرکهای سمپاتیک و داروهای مرتبط با پروستاگلاندین (PG) مؤثر هستند. بریمونیدین (محرک گیرنده آلفا آدرنرژیک) در نوزادان به دلیل خطر سرکوب سیستم عصبی مرکزی در کودکان زیر ۲ سال منع مصرف دارد. در صورت نگرانی از آسیب اپیتلیوم قرنیه، از فرآوردههای بدون مواد نگهدارنده استفاده کنید.
جراحی بازسازی مسیر خروجی (گونیوتومی و ترابکولوتومی): به عنوان جراحی اولیه توصیه میشود16). گزارشهایی از گونیوتومی پیشگیرانه نیز وجود دارد. با این حال، در مواردی که عنبیه باقیمانده ترابکولوم را میپوشاند، ممکن است بیاثر باشد.
جراحی فیلتراسیون (ترابکولکتومی): فقط گزارشهای محدود و کوتاهمدت تا میانمدت وجود دارد. در چشم کودکان تمایل به نتایج ضعیف دارد و فیستول پس از جراحی در حدود ۲۵٪ موارد رخ میدهد13). همچنین گزارشهایی از گلوکوم بدخیم پس از جراحی وجود دارد.
جراحی ایمپلنت گلوکوم (جراحی شنت لولهای): دستگاههای Baerveldt و Ahmed قابل استفاده هستند. در چشمهای دارای لنز طبیعی، توصیه میشود لوله به صورت مماسی وارد شود نه به سمت مرکز قرنیه. کنترل خوب فشار داخل چشم قابل انتظار است.
سیکلوکواگولاسیون: آخرین راهکار. گزارش شده است که سیکلوکریوتراپی در بسیاری از موارد منجر به فیستول میشود. به دلیل هیپوپلازی جسم مژگانی، خطر فیستول در مقایسه با چشم سالم بیشتر است.
به دلیل ناهنجاری رشدی زاویه، رویکرد متفاوتی نسبت به گلوکوم زاویه باز معمولی لازم است. اولین گزینه جراحی بازسازی مسیر خروجی است و سپس جراحی شنت لوله گزینه مناسبی است. بریمونیدین در کودکان زیر ۲ سال منع مصرف دارد و استفاده از داروهای ضد متابولیت ممکن است کراتوپاتی آنیریدیا را تشدید کند، بنابراین نیاز به قضاوت دقیق دارد8).
مراقبت از کمبینایی باید از مراحل اولیه آغاز شود8).
اصلاح عیوب انکساری اساسی است و میزان همراهی نزدیکبینی ۶۴٪ یا بیشتر گزارش شده است.
درمان فوتوفوبیا برای حفظ رشد بینایی و کیفیت زندگی مهم است8).
بیشتر بیماران میتوانند در مدارس عادی تحصیل کنند، اما به کمکهایی مانند کتابهای درسی بزرگنما، تبلت و پایه کتاب نیاز دارند. شرکت در کلاسهای ویژه کمبینایان یا استفاده از مشاورههای تربیتی و تحصیلی مدارس نابینایان و مدارس ویژه حمایت از بینایی نیز گزینههایی هستند.
از آوریل ۲۰۱۷، این بیماری به عنوان یک بیماری نادر مشخص تعیین شده است، بنابراین حتی اگر کارت معلولیت جسمی دریافت نکرده باشید، در صورت شدت درجه III یا بالاتر، مشمول کمک هزینه پزشکی و تأمین وسایل کمککننده خواهید بود7). وسایل کمککننده شامل عینک طبی، عینک ضد نور، لنزهای تماسی (از جمله لنزهای دارای عنبیه مصنوعی)، عینک مخصوص کمبینایان، عصای ایمنی نابینایان و چشم مصنوعی میشود.
PAX6 یک DNA ژنومی به طول 22 کیلوباز شامل 14 اگزون است که 422 اسید آمینه را کد میکند1). دارای دو دامنه اتصال به DNA (دامنه جفتی و هومئودومین جفتی) است و دامنه PST (غنی از پرولین، سرین و ترئونین) در انتهای C به عنوان فعالکننده رونویسی عمل میکند.
PAX6 تکثیر، تمایز، مهاجرت و چسبندگی سلولها را تنظیم میکند و اهداف آن شامل خود PAX6 و همچنین ژنهای کدکننده کریستالین عدسی و کراتین قرنیه است. بیان آن در شبکیه، عدسی و قرنیه بالغ ادامه مییابد. ژن PAX6 یکی از ژنهای کنترل اصلی است که تمایز اندامها را در دوره جنینی هدایت میکند.
بیشتر جهشهای PAX6 از طریق تخریب mRNA وابسته به جهش بیمعنی (NMD) باعث نارسایی هاپلوئیدی میشوند1). جهشهایی که کدون توقف زودرس (PTC) ایجاد میکنند (جهشهای بیمعنی، جهشهای تغییر چارچوب و بیشتر جهشهای پیرایش) منجر به فنوتیپ کلاسیک آنیریدیا میشوند.
از سوی دیگر، اگر PTC در اگزون نهایی یا در ۵۰ جفت باز انتهایی اگزون ماقبل آخر قرار گیرد، از NMD فرار کرده و پروتئین کوتاهشده ترجمه میشود که میتواند منجر به فنوتیپ شدید شود1).
یک مورد نادر از همراهی جهش nonsense PAX6 با c.282C>A (p.Cys94*) و تریزومی ۲۱ در یک بیمار گزارش شده است. جهش PAX6 به صورت de novo رخ داده و بیمار آنیریدی دوطرفه کامل، گلوکوم مادرزادی، AAK و هیپوپلازی فووه آ را نشان داد2).
اگرچه همبستگی قطعی ژنوتیپ-فنوتیپ ثابت نشده است، برخی تمایلات شناخته شدهاند1).
در سری گونیوسکوپی گرانت و والتون نشان داده شد که در مراحل اولیه، استرومای عنبیه به سمت جلو بر روی ترابکول کشیده شده و اتصال چسبندهای ایجاد میکند که به تدریج به صورت لایهای درآمده و نهایتاً منجر به انسداد زاویه میشود14). این مکانیسم عامل اصلی ایجاد گلوکوم است. از نظر پاتولوژیک، فقدان عضله صاف با حفظ ریشه عنبیه و ناهنجاری رشدی زاویه زمینهساز آن است.
AAK عمدتاً توسط کمبود سلولهای بنیادی لیمبوس (LSCD) ایجاد میشود، اما تمایز غیرطبیعی اپیتلیوم قرنیه، چسبندگی غیرطبیعی، نفوذ سلولهای ملتحمه و تولید ناکافی اشک نیز نقش دارند. کمبود ماتریکس متالوپروتئیناز ۹ (MMP-9) که توسط PAX6 تنظیم میشود، منجر به تجمع فیبرین و نفوذ سلولهای التهابی میشود و بههمریختگی توالی کلاژن در استروما باعث از دست رفتن شفافیت میگردد.
AAK به پنج مرحله تقسیم میشود. در مرحله I فقط ناهنجاری اپیتلیوم محیطی، در مرحله II تغییرات اپیتلیال گریز از مرکز (بدون رسیدن به مرکز)، در مرحله III تغییرات اپیتلیال در قرنیه مرکزی و نئوواسکولاریزاسیون سطحی محیطی، در مرحله IV نئوواسکولاریزاسیون سطحی تمام قرنیه، و در مرحله V ناهنجاری اپیتلیال تمام قرنیه و اسکار عمقی استروما مشاهده میشود10).
بین وضعیت جهش PAX6 و پیشرفت AAK ارتباط وجود دارد. در بیماران دارای جهش PTC یا طویلشدگی C-ترمینال، AAK به صورت وابسته به سن پیشرفت میکند، در حالی که در سایر انواع جهش ممکن است کراتوپاتی غیرپیشرونده مشاهده شود11).
سندرم گیلِسپی در اثر جهش در ژن ITPR1 ایجاد میشود 3). ITPR1 عضوی از خانواده گیرندههای IP3 است که کانالهای آزادکننده Ca²⁺ را تشکیل میدهد و در شبکه آندوپلاسمی موضعی میشود. جهشهای غالب-منفی بر تشکیل و نگهداری عضله اسفنکتر عنبیه تأثیر میگذارند و منجر به دیسژنزی خاص عنبیه در اطراف مردمک و میدریاز ثابت میشوند.
در مرور ادبیات سندرم گیلِسپی توسط Ciaccio و همکاران (2024)، تجزیه و تحلیل 33 مورد تأیید شده مولکولی نشان داد که رشد حرکتی با تأخیر همراه است اما در طول زمان بهبود مییابد، ناتوانی ذهنی در همه موارد وجود ندارد و 17% هوش طبیعی دارند، و علائم عصبی غیرپیشرونده هستند 3).
با گسترش فناوری توالییابی کل اگزوم، جهشهای جدید PAX6 به طور مداوم شناسایی میشوند. تا سال ۲۰۱۸، ۴۹۱ جهش در پایگاه داده جهشهای PAX6 انسانی ثبت شده بود و از آن زمان تاکنون حدود ۲۵۰ جهش جدید گزارش شده است1). مواردی از جهش در نواحی غیرکدکننده که باعث آنیریدیا میشوند نیز در حال شناسایی هستند و انتظار میرود مواردی که با آزمایشهای قبلی قابل تشخیص نبودند، روشن شوند9).
در جراحی آب مروارید در موارد همراه با AAK شدید، تکنیک تجسم با کمک نورپردازی معکوس لوستر (chandelier) مفید است4). این روش امکان فیکوامولسیفیکاسیون ایمن را حتی در بیماران با AAK درجه ۳ تا ۴ فراهم کرده و بهبود بینایی پس از جراحی حاصل شده است.
نوع جهش PAX6 در حال آشکار شدن است که الگوی پیشرفت AAK را متفاوت میکند. با کاهش هزینه آزمایشهای ژنتیکی، پیشبینی سیر بالینی بر اساس نوع جهش و مداخله زودهنگام به گزینهای واقعی تبدیل شده است.
در موارد ترکیبی آنیریدیا و تریزومی ۲۱، مواردی گزارش شده است که با وجودهمزمانی دو بیماری، سیر نسبتاً خفیفی داشتهاند2). درک تأثیرهمزمانی اختلالات ژنتیکی متعدد در یک بیمار بر فنوتیپ، میتواند بینش مهمی برای پزشکی فردمحور در آینده فراهم کند.
کاربرد داروی read-through (ataluren) برای جهشهای نوع PTC در آنیریدیا در سطح تحقیقات پایه بررسی شده است8). در مورد ژن درمانی PAX6، تحقیقات پایه در حال انجام است که با استفاده از ناقل AAV-PAX6 در مدل موش جهشیافته Sey، جایگزینی ژن را انجام میدهد. انتظار میرود که این تحقیقات به کارآزماییهای بالینی در آینده منجر شود.
کارآزماییهای بالینی پیوند ورقههای سلولهای اپیتلیال قرنیه مشتق از سلولهای بنیادی پرتوان القایی (iPS) در داخل و خارج از کشور در حال انجام است و به عنوان یک روش درمانی جدید برای AAK مورد توجه قرار گرفته است 8). عنبیه مصنوعی (مانند CustomFlex Artificial Iris) در خارج از کشور سابقه استفاده انباشته دارد. لنزهای تماسی با عنبیه مصنوعی به عنوان وسایل کمکدرمانی تحت پوشش بیمه هستند.
شناسایی وضعیت واقعی از طریق انباشت دادههای رجیستری در مقیاس بزرگ در ژاپن و بهبود کیفیت شواهد، از مسائل مهم آینده است 8). پیشبینی پیشرفت AAK بر اساس جهشهای ژنتیکی فردی و بهینهسازی مداخله زودهنگام مورد انتظار است.