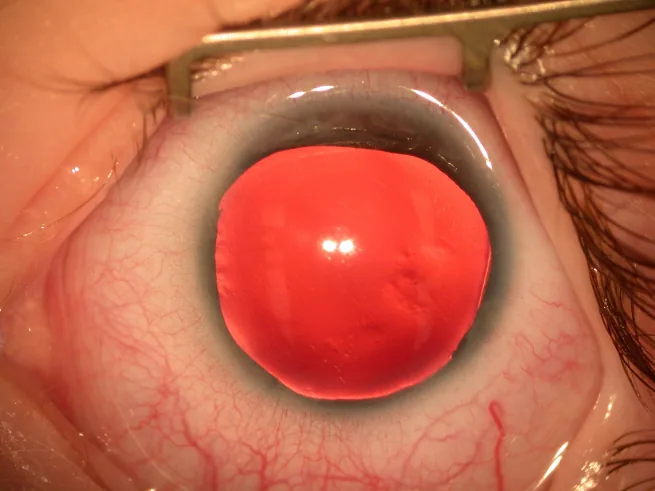
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
Wang Q, Wei WB, Shi XY, Rong WN.. A novel PAX6 variant as the cause of aniridia in a Chinese patient with SRRRD. BMC Med Genomics. 2023;16(1):182. doi:10.1186/s12920-023-01620-w. PMID:37542296; PMCID:PMC10401864.
Vasilyeva TA, Sukhanova NV, Marakhonov AV, Kuzina NY, Shilova NV, Kadyshev VV, et al. Co-Occurrence of Congenital Aniridia Due to Nonsense PAX6 Variant p.(Cys94*) and Chromosome 21 Trisomy in the Same Patient. International journal of molecular sciences. 2023;24(21). doi:10.3390/ijms242115527. PMID:37958513; PMCID:PMC10650867.
Ciaccio C, Taddei M, Pantaleoni C, Grisoli M, Di Bella D, Magri S, Taroni F, D’Arrigo S. Phenotypic Spectrum and Natural History of Gillespie Syndrome. An Updated Literature Review with 2 New Cases. Cerebellum (London, England). 2024;23(6):2655-2670. doi:10.1007/s12311-024-01733-7. PMID:39177731; PMCID:PMC11585489.
Hu J, Hu CC. Chandelier retroillumination-assisted cataract surgery in two cases of congenital aniridia with severe aniridia-associated keratopathy: case series. Therapeutic advances in ophthalmology. 2024;16:25158414241302879. doi:10.1177/25158414241302879. PMID:39619855; PMCID:PMC11605765.
Christodoulou E, Batsos G, Parikakis E, Papadopoulos V, Karagiannis D, Koumoutsos PP, et al. Combined Post-Traumatic Total Aniridia and Glaucoma Management. Case reports in ophthalmology. 2021;12(1):204-207. doi:10.1159/000511099. PMID:33976683; PMCID:PMC8077530.
Ratna R, Tibrewal S, Gour A, Gupta R, Mathur U, Vanita V. A rare case of congenital aniridia with an unusual run-on mutation in PAX6 gene. Indian journal of ophthalmology. 2022;70(7):2661-2664. doi:10.4103/ijo.IJO_433_22. PMID:35791194; PMCID:PMC9426074.
Lim HT, Kim DH, Kim H. PAX6 aniridia syndrome: clinics, genetics, and therapeutics. Current opinion in ophthalmology. 2017;28(5):436-447. doi:10.1097/ICU.0000000000000405. PMID:28598868.
Edén U, Riise R, Tornqvist K.. Corneal involvement in congenital aniridia. Cornea. 2010;29(10):1096-1102. doi:10.1097/ico.0b013e3181d20493. PMID:20567200.
Nishida K, Kinoshita S, Ohashi Y, Kuwayama Y, Yamamoto S.. Ocular surface abnormalities in aniridia. Am J Ophthalmol. 1995;120(3):368-375. doi:10.1016/s0002-9394(14)72167-1. PMID:7661209.
Holland EJ, Djalilian AR, Schwartz GS. Management of aniridic keratopathy with keratolimbal allograft: a limbal stem cell transplantation technique. Ophthalmology. 2003;110(1):125-30. doi:10.1016/s0161-6420(02)01451-3. PMID:12511357.
Wiggins RE, Tomey KF.. The results of glaucoma surgery in aniridia. Arch Ophthalmol. 1992;110(4):503-505. doi:10.1001/archopht.1992.01080160081036. PMID:1562257.
Grant WM, Walton DS. Progressive changes in the angle in congenital aniridia, with development of glaucoma. American journal of ophthalmology. 1974;78(5):842-7. doi:10.1016/0002-9394(74)90308-0. PMID:4423758.
Tsai JH, Freeman JM, Chan CC, Schwartz GS, Derby EA, Petersen MR, et al. A progressive anterior fibrosis syndrome in patients with postsurgical congenital aniridia. American journal of ophthalmology. 2005;140(6):1075-9. doi:10.1016/j.ajo.2005.07.035. PMID:16376654.
Adachi M, Dickens CJ, Hetherington J Jr, Hoskins HD, Iwach AG, Wong PC, et al. Clinical experience of trabeculotomy for the surgical treatment of aniridic glaucoma. Ophthalmology. 1997;104(12):2121-5. doi:10.1016/s0161-6420(97)30041-4. PMID:9400774.
Vincent SJ. The use of contact lenses in low vision rehabilitation: optical and therapeutic applications. Clinical & experimental optometry. 2017;100(5):513-521. doi:10.1111/cxo.12562. PMID:28664572.