孤立性無虹彩症
頻度:全体の約2/3。
遺伝形式:常染色体優性(AD)。
特徴:PAX6遺伝子変異による。全身症状を伴わない。浸透率は完全だが表現度は多様。

無虹彩症(Aniridia)は、虹彩のさまざまな程度の形成不全または欠損を特徴とする稀な先天性疾患である。「無虹彩症」の名は誤称であり、隅角鏡検査や超音波生体顕微鏡(UBM)では虹彩組織の断片がほぼ常に確認される。
有病率は約1/40,000〜1/100,000とされ、顕著な人種差や性差は報告されていない1)。ICD-10ではQ13.1に分類される。
虹彩だけでなく、角膜・水晶体・隅角・中心窩・視神経にも影響が及ぶ汎眼球性疾患であり1)、視力を脅かす多彩な眼合併症を呈する。視力予後はおおむね不良で、矯正視力0.1程度にとどまることが多い。瞳孔反応は消失しているが調節反応は保たれており、60〜90%が両眼性である。
以下の3つの表現型が認められている。
孤立性無虹彩症
頻度:全体の約2/3。
遺伝形式:常染色体優性(AD)。
特徴:PAX6遺伝子変異による。全身症状を伴わない。浸透率は完全だが表現度は多様。
WAGR症候群
頻度:散発例の一部。
遺伝形式:PAX6とWT1の隣接欠失。
特徴:Wilms腫瘍・泌尿生殖器異常・精神遅滞を合併。腫瘍リスクは最大50%。
ギレスピー症候群
頻度:全体の約2%。
遺伝形式:ITPR1遺伝子変異。
特徴:小脳失調・知的障害を伴う。固定散瞳を呈する特異的な虹彩異常が特徴的3)。
散発性の無虹彩症は全体の約1/3を占め、PAX6を含む11p13のde novo欠失によって生じる。隣接するWT1遺伝子にまで欠失が及ぶ場合はWAGR症候群の原因となる1)。散発性無虹彩症の25〜30%がWilms腫瘍を発症し、相対リスクは67と報告されている。
PAX6は眼形成のマスターコントロール遺伝子であり、眼・神経管・嗅球・膵臓Langerhans島・嗅上皮の発達に関与する。片アリルの機能喪失(ハプロ不全)で発症し、両アリルが異常の場合には胎生致死となる。2017年に難病法の定める指定難病となり、重症度Ⅲ度以上(詳細は診断・検査の項参照)で医療費助成の対象となる7)。
散発性(新規変異)は全体の約1/3を占め、家族歴がなくても発症する。散発例ではWAGR症候群の可能性があるため、遺伝子検査と腹部超音波によるWilms腫瘍スクリーニングが重要である。
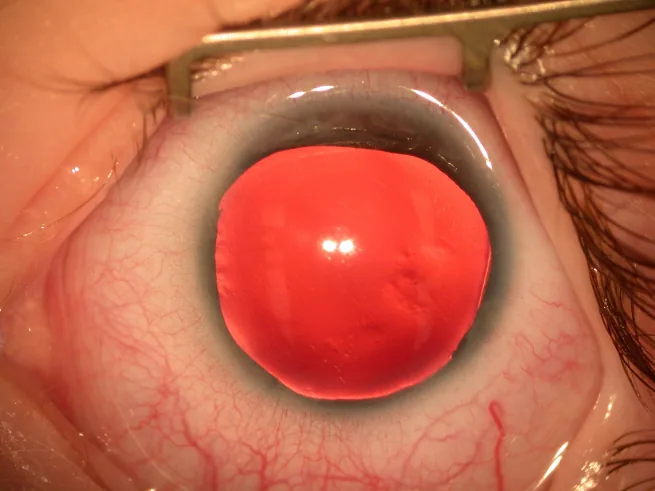
無虹彩症の多くは出生時の虹彩・瞳孔異常、または乳児期の眼振によって発見される。
表現型は家族間および家族内で異なるが、左右眼での差は通常小さい。
中心窩形成不全が主因となり、矯正視力は0.1〜0.2程度のことが多い。黄斑低形成を合併した場合は特に視力予後が不良である。乳幼児期からの屈折矯正とロービジョンケアが視覚発達に重要である。
PAX6は眼組織のほか中枢神経・膵臓Langerhans島・嗅上皮にも発現するため、以下の眼外合併症を認める場合がある8)。
視機能を決める重要因子は緑内障・黄斑低形成・眼球振盪症・角膜症・白内障・虹彩形成異常であり、緑内障による視野・視力障害は不可逆的であるため経過観察において眼圧管理が最重要である8)。
大多数の先天無虹彩症は、第11染色体短腕(11p13)に位置するPAX6遺伝子のヘテロ接合型変異によって生じる。PAX6はハプロ不全が主な発症メカニズムである1)。
PAX6遺伝子は眼形成のマスターコントロール遺伝子であり、眼・神経管・嗅球・膵臓の発達に重要な役割を果たす。正常な眼の発達には2コピーのPAX6が必要であり、片方の機能喪失だけで無虹彩症が発症する1)。
中国人患者のコホート研究では、96.9%の症例でPAX6遺伝子に原因変異が同定されている1)。典型的な無虹彩症ではナンセンス変異依存mRNA分解(NMD)を誘導する変異または大規模欠失が96%で検出される1)。
病理学的には虹彩根部を残して平滑筋が欠損しており、隅角の発達不全がみられる。角膜上皮幹細胞の機能異常がみられ、上皮とBowman膜に異常をきたし、血管豊富なパンヌスが形成される。
無虹彩症の表現型を引き起こすPAX6変異の内訳を以下に示す。
| 変異タイプ | 頻度 |
|---|---|
| ナンセンス変異 | 約39% |
| フレームシフト変異 | 約25% |
| スプライス変異 | 約13% |
| ミスセンス変異 | 約12% |
ランオン変異(C末端延長変異)は約5%を占め、ストップコドンが翻訳コドンに変換されることで異常に延長したPAX6タンパク質が産生される6)。C末端延長変異は重症の虹彩形成不全と高度な視力障害を伴うことが多い1)6)。
遺伝子変異はPTC型変異が多く、ミスセンス変異の報告もある7)。遺伝学的検査の有用性について、SangerシークエンシングまたはNGSでisolated aniridiaの85%近くに変異が検出される。加えてMLPAまたはCMAでPAX6遺伝子内またはcis-regulatory領域の欠損が15%近くに検出される8)。
Wang(2023)はフレームシフト変異c.640_646del(p.R214Pfs*28)を新規同定し、完全な虹彩欠損・中心窩形成不全・水晶体偏位・網膜剥離を呈した症例を報告した1)。
Ratnaら(2022)はインド人家系においてランオン変異c.1268A>T(p.*423L)を同定した。罹患者は完全無虹彩・眼振・中心窩形成不全・AAK・水晶体上方亜脱臼・強度近視・視神経萎縮を呈し、C末端延長変異による重症表現型を示した6)。
散発性無虹彩症では、PAX6に加えてWT1遺伝子を含む大規模欠失がWAGR症候群の原因となる。WT1欠失を有する場合のWilms腫瘍リスクは最大50%である1)。WAGR症候群の疑いがあれば遺伝学的検査でPAX6とWT1の欠損を確認し、Wilms腫瘍リスク評価と発達遅延の経過観察が可能となる8)。遺伝子検査によるWT1領域の評価が不可欠であり、散発性の孤発例の30%は5歳までにWilms腫瘍を発症するとされる。WT1遺伝子がPAX6の近傍に位置することから、両者がともに欠損する第11染色体短腕欠失(11p13欠失)で無虹彩症にWilms腫瘍が合併する。
ギレスピー症候群はITPR1遺伝子のヘテロ接合型ドミナントネガティブ変異または両アレル変異によって生じる3)。これまでに分子診断が確認された症例は37例と報告されており、Gly2554残基がホットスポットとして知られる3)。
無虹彩症の診断基準(2020年)に基づき、以下の基準で確定する7)。
A. 症状
B. 検査所見
C. 鑑別すべき疾患
E. 遺伝学的検査:PAX6遺伝子の病的変異もしくは11p13領域の欠失
診断カテゴリー7):
難病認定のための重症度分類は以下の4段階で規定されている7)。
| 重症度 | 定義 |
|---|---|
| Ⅰ度 | 片眼罹患、僚眼健常 |
| Ⅱ度 | 両眼罹患、良好な方の矯正視力0.3以上 |
| Ⅲ度 | 両眼罹患、良好な方の矯正視力0.1以上0.3未満 |
| Ⅳ度 | 両眼罹患、良好な方の矯正視力0.1未満 |
Ⅰ〜Ⅲ度であっても、緑内障等による視野狭窄(Goldmann I/4視標で中心残存視野20度以内)を伴う場合は1段階上の重症度に移行する。重症度Ⅲ度以上が医療費助成の対象となる7)。
細隙灯顕微鏡により虹彩の欠損または形成不全を確認すれば臨床診断は容易である。隅角鏡検査や超音波生体顕微鏡で残存虹彩組織の評価を行う。前房隅角の発達異常の有無も確認する。
以下の眼合併症を系統的に評価する。
無虹彩症の遺伝学的評価において最も重要な目標は、PAX6欠失がWT1遺伝子にまで及ぶかどうかの確認である1)。全エクソーム解析やMLPA法によりPAX6およびWT1領域の変異・欠失を評価する1)2)。
散発性無虹彩症ではWT1遺伝子の欠失によるWilms腫瘍リスクの評価が生命予後に直結する1)。家族性であっても表現型の多様性があるため、遺伝子検査による確定診断と遺伝カウンセリングが推奨される。
無虹彩症に対する根本的治療法は存在しない。残存視力を最大限活用するロービジョンケアと、各合併症に対する個別の治療が管理の中心となる8)。
角膜実質混濁に対する角膜移植は慎重に判断する8)。
角膜移植により短期的に視力改善が得られる場合があるが、黄斑低形成等の併発症により改善は限定的である。長期的には緑内障の進行および移植片機能不全により視力予後は不良となる。
角膜上皮幹細胞疲弊症では手術加療を検討する8)。
白内障手術は、混濁と羞明の程度をみて検討する8)。
白内障は20歳までに50〜85%で発症する。混濁と羞明の強度により手術を計画する。手術症例の66〜100%で視力改善がみられたと報告されているが、以下の点に注意が必要である。
Zinn小帯が脆弱であることから、眼内レンズ挿入は慎重な適応となる。
Huら(2024)は重度のAAKを伴う先天無虹彩症2例に対し、シャンデリア逆照明補助下での水晶体乳化吸引術を施行した。角膜混濁のため通常の術中可視化が困難であったが、後方からの照明により水晶体と前嚢の明瞭な可視化が可能となり、術後3週で矯正視力がそれぞれ20/200および20/1000に改善した4)。
緑内障は視機能予後に直結するため、積極的に治療する8)。
緑内障発症後は以下の5段階アルゴリズムに基づいて管理する。
薬物療法:β遮断薬・交感神経刺激薬・プロスタグランジン(PG)関連薬が有効。乳幼児へのブリモニジン(αアドレナリン受容体刺激薬)は中枢神経抑制リスクがあるため2歳未満には禁忌。角膜上皮障害が懸念される場合は防腐剤を含まない製剤を使用する。
流出路再建術(隅角切開術・線維柱帯切開術):初回手術として推奨16)。予防的隅角切開術の報告もある。ただし残存虹彩が線維柱帯を覆う症例では無効となる場合がある。
濾過手術(線維柱帯切除術):少数例・中短期の報告にとどまる。小児眼では成績不良な傾向があり、術後の眼球ろうは約25%に及ぶ13)。術後悪性緑内障の報告もあり。
緑内障インプラント手術(チューブシャント手術):Baerveldt・Ahmed型デバイスが使用可能。有水晶体眼へのチューブ挿入は角膜中央方向でなく接線方向へを推奨。良好な眼圧コントロールが期待できる。
毛様体凝固術:最終手段。毛様体冷凍凝固では多くが眼球ろうに陥ったとの報告がある。毛様体低形成があるため健常眼より眼球ろうリスクが高い。
隅角の発達異常が背景にあるため、通常の開放隅角緑内障とは異なるアプローチが必要である。初回は流出路再建術を選択し、次いでチューブシャント手術が良好な選択肢となる。ブリモニジンは2歳未満には使用禁忌であり、抗代謝薬の使用はAAKを悪化させうるため慎重な判断が求められる8)。
ロービジョンケアは早期から導入する8)。
屈折矯正が基本であり、近視の合併率は64%以上とされる。
羞明治療は視機能発達と生活の質を保つために重要である8)。
患者の多くは普通学級に進学可能であるが、拡大教科書・タブレット端末・書見台などの支援が必要となる。弱視学級への通級や、盲学校・視覚特別支援学校による育児相談・教育相談の利用も選択肢となる。
2017年4月より指定難病に認定されたため、身体障害者手帳を取得していない場合でも、重症度Ⅲ度以上であれば医療費助成・補装具支給の対象となる7)。対象となる補装具には、矯正眼鏡、遮光眼鏡、コンタクトレンズ(人工虹彩付き含む)、弱視眼鏡、視覚障害者安全つえ、義眼が含まれる。
PAX6は14個のエキソンを含む22kbのゲノムDNAに及び、422個のアミノ酸をコードする1)。2つのDNA結合ドメイン(ペアードドメインとペアード型ホメオドメイン)を有し、C末端のPST(プロリン・セリン・スレオニンに富む)ドメインが転写活性化因子として機能する。
PAX6は細胞の増殖・分化・移動・接着を制御し、その標的にはPAX6自身のほか、水晶体クリスタリンや角膜ケラチンをコードする遺伝子が含まれる。成人の網膜・水晶体・角膜でも発現が継続する。PAX6遺伝子は胎生期の器官分化を司るマスターコントロール遺伝子の一つである。
ほとんどのPAX6変異はナンセンス変異依存mRNA分解(NMD)を介してハプロ不全を引き起こす1)。早期終止コドン(PTC)を導入する変異(ナンセンス変異・フレームシフト変異・ほとんどのスプライス変異)が典型的な無虹彩症表現型をもたらす。
一方、PTC が最終エキソンまたは最後から2番目のエキソンの末端50bp以内に位置する場合はNMDを逃れ、切断型タンパク質が翻訳されて重症表現型を呈する可能性がある1)。
PAX6のナンセンス変異c.282C>A(p.Cys94*)と21トリソミーが同一患者に合併した稀な症例が報告されている。PAX6変異はde novoに生じ、完全両眼無虹彩・先天緑内障・AAK・中心窩形成不全を呈した2)。
明確な遺伝子型–表現型相関は確立していないものの、いくつかの傾向が知られている1)。
GrantとWaltonの隅角鏡検査シリーズでは、初期に虹彩実質が線維柱帯上に前方伸展して癒着様の付着を形成し、次第にシート状となり最終的に隅角閉塞に至ることが示された14)。このメカニズムが緑内障発症の主要因である。病理学的には虹彩根部を残した平滑筋の欠損と隅角の発達不全が基盤をなす。
AAKは主に輪部幹細胞欠損(LSCD)によって引き起こされるが、角膜上皮の異常分化・接着異常・結膜細胞の浸潤・涙液産生不足も関与する。PAX6によって調節されるマトリックスメタロプロテアーゼ9(MMP-9)の欠乏がフィブリン蓄積と炎症細胞浸潤を引き起こし、実質のコラーゲン配列の乱れから透明性が失われる。
AAKは5段階に分類される。Stage Iでは周辺上皮のみの異常、Stage IIでは求心性上皮変化(中心部は未到達)、Stage IIIでは中心角膜の上皮変化と周辺の表層新生血管、Stage IVでは全角膜の表層新生血管、Stage Vでは全角膜の上皮異常と深層実質瘢痕を呈する10)。
PAX6変異ステータスとAAKの進行には関連がある。PTCやC末端延長変異を有する患者ではAAKが年齢依存性に進行する一方、他の変異型では非進行性の角膜症を呈することがある11)。
ギレスピー症候群はITPR1遺伝子の変異によって生じる3)。ITPR1はIP3受容体ファミリーの一員でCa²⁺放出チャネルを形成し、小胞体に局在する。ドミナントネガティブ変異は虹彩括約筋の形成・維持に影響を与え、瞳孔周囲の特異的な虹彩形成不全と固定散瞳をもたらす。
Ciaccioら(2024)のギレスピー症候群文献レビューでは、分子確認された33例の解析から、運動発達は遅延するものの経時的に改善すること、知的障害は全例には認められず17%で正常知能であること、神経学的徴候は非進行性であることが確認された3)。
全エクソーム解析技術の普及により、新規PAX6変異の同定が続いている。Human PAX6 Mutation Databaseの2018年時点で491変異が登録され、以降も約250の新規変異が報告されている1)。ノンコーディング領域の変異が無虹彩症の原因となる事例も同定されつつあり、従来の検査では診断できなかった症例の解明が期待される9)。
重度のAAKを伴う症例での白内障手術には、シャンデリア逆照明補助による可視化技術が有用である4)。この手技はGrade 3〜4の高度AAKを有する患者においても安全な水晶体乳化吸引術を可能にし、術後視力改善が得られている。
PAX6変異の種類によってAAKの進行パターンが異なることが明らかになりつつある。遺伝子検査のコスト低下により、変異型に基づく臨床経過の予測と早期介入が現実的な選択肢となりつつある。
無虹彩症と21トリソミーの合併例では、両疾患の共存にもかかわらず比較的軽症の経過を示した例が報告されている2)。複数の遺伝的障害が同一患者に共存した場合の表現型への影響を理解することは、今後の個別化医療に重要な知見をもたらす可能性がある。
PTC型変異に対するリードスルー薬(ataluren)の無虹彩症への応用が基礎研究レベルで検討されている8)。PAX6遺伝子治療については、Sey変異マウスモデルを用いたAAV-PAX6ベクターによる遺伝子補充の基礎研究が進行中である。今後の臨床試験への展開が期待される。
iPS細胞由来角膜上皮細胞シート移植の治験が国内外で実施されており、AAKに対する新たな治療法として注目される8)。人工虹彩(CustomFlex Artificial Iris等)については海外での使用実績が蓄積されている。補装具として人工虹彩付きコンタクトレンズは保険適用の対象である。
本邦における大規模レジストリデータの蓄積による実態把握と、エビデンスの質向上が今後の重要課題である8)。個々の遺伝子変異に基づくAAKの進行予測と早期介入の最適化が期待される。