Aniridia Isolada
Frequência: cerca de 2/3 do total.
Padrão de herança: autossômico dominante (AD).
Características: causada por mutação no gene PAX6. Sem sintomas sistêmicos. Penetrância completa, mas expressividade variável.

Aniridia é uma doença congênita rara caracterizada por formação incompleta ou ausência da íris em vários graus. O termo “aniridia” é um equívoco, pois fragmentos de tecido iridiano são quase sempre detectáveis por gonioscopia ou ultrassonografia biomicroscópica (UBM).
A prevalência é de aproximadamente 1/40.000 a 1/100.000, sem diferenças raciais ou de gênero significativas relatadas 1). Na CID-10, é classificada como Q13.1.
Trata-se de uma doença pan-ocular que afeta não apenas a íris, mas também a córnea, o cristalino, o ângulo da câmara anterior, a fóvea e o nervo óptico 1), apresentando diversas complicações oculares que ameaçam a visão. O prognóstico visual é geralmente ruim, com acuidade visual corrigida frequentemente em torno de 0,1. O reflexo pupilar está ausente, mas a acomodação é preservada, e 60-90% dos casos são bilaterais.
Os três fenótipos a seguir são reconhecidos.
Aniridia Isolada
Frequência: cerca de 2/3 do total.
Padrão de herança: autossômico dominante (AD).
Características: causada por mutação no gene PAX6. Sem sintomas sistêmicos. Penetrância completa, mas expressividade variável.
Síndrome de WAGR
Frequência: parte dos casos esporádicos.
Padrão de herança: deleção contígua de PAX6 e WT1.
Características: associada a tumor de Wilms, anomalias geniturinárias e retardo mental. Risco de tumor de até 50%.
Síndrome de Gillespie
Frequência: cerca de 2% do total.
Padrão de herança: mutação no gene ITPR1.
Características: acompanhada de ataxia cerebelar e deficiência intelectual. Apresenta anomalia iriana característica com midríase fixa3).
A aniridia esporádica representa cerca de 1/3 do total e é causada por deleção de novo em 11p13, incluindo PAX6. Quando a deleção se estende ao gene WT1 adjacente, causa a síndrome de WAGR1). 25 a 30% dos casos de aniridia esporádica desenvolvem tumor de Wilms, com risco relativo relatado de 67.
PAX6 é o gene mestre do desenvolvimento ocular, envolvido no desenvolvimento do olho, tubo neural, bulbo olfatório, ilhotas de Langerhans do pâncreas e epitélio olfatório. A doença ocorre por perda de função de um alelo (haploinsuficiência); a anormalidade bialélica é letal embrionária. Em 2017, foi designada doença intratável pela lei de doenças intratáveis, sendo elegível para subsídio de custos médicos para gravidade grau III ou superior (consulte a seção de diagnóstico e exames para detalhes)7).
Casos esporádicos (nova mutação) representam cerca de 1/3 do total e podem ocorrer sem histórico familiar. Em casos esporádicos, há possibilidade de síndrome de WAGR, sendo importante realizar teste genético e ultrassonografia abdominal para rastreamento de tumor de Wilms.
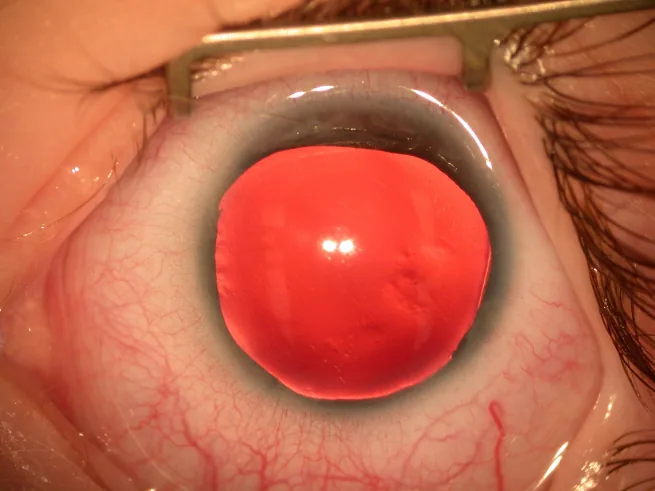
A maioria dos casos de aniridia é descoberta ao nascimento por anomalias da íris e pupila, ou na infância devido ao nistagmo.
O fenótipo varia entre famílias e dentro da mesma família, mas a diferença entre os olhos direito e esquerdo costuma ser pequena.
Devido principalmente à hipoplasia foveal, a acuidade visual corrigida é frequentemente de 0,1 a 0,2. O prognóstico visual é particularmente ruim quando há hipoplasia macular associada. A correção refrativa desde a infância e os cuidados de baixa visão são importantes para o desenvolvimento visual.
O PAX6 é expresso não apenas nos tecidos oculares, mas também no sistema nervoso central, nas ilhotas de Langerhans do pâncreas e no epitélio olfatório, podendo apresentar as seguintes complicações extraoculares 8).
Os fatores importantes que determinam a função visual são glaucoma, hipoplasia macular, nistagmo, ceratopatia, catarata e anomalias da íris. Como os danos ao campo visual e à acuidade visual causados pelo glaucoma são irreversíveis, o controle da pressão intraocular é fundamental no acompanhamento.8)
A maioria dos casos de aniridia congênita é causada por mutações heterozigóticas no gene PAX6, localizado no braço curto do cromossomo 11 (11p13). A haploinsuficiência do PAX6 é o principal mecanismo de desenvolvimento da doença.1)
O gene PAX6 é um gene mestre controlador da formação ocular, desempenhando um papel crucial no desenvolvimento dos olhos, tubo neural, bulbo olfatório e pâncreas. Duas cópias do PAX6 são necessárias para o desenvolvimento ocular normal, e a perda de função de apenas uma cópia já causa aniridia.1)
Em um estudo de coorte com pacientes chineses, mutações causais no gene PAX6 foram identificadas em 96,9% dos casos.1) Na aniridia típica, mutações que induzem degradação do mRNA dependente de mutação sem sentido (NMD) ou deleções de grande escala são detectadas em 96% dos casos.1)
Patologicamente, há ausência de músculo liso, restando apenas a raiz da íris, e observa-se desenvolvimento anormal do ângulo. Há disfunção das células-tronco epiteliais da córnea, levando a anormalidades no epitélio e na membrana de Bowman, com formação de pannus vascularizado.
Abaixo estão as distribuições das mutações no PAX6 que causam o fenótipo de aniridia.
| Tipo de mutação | Frequência |
|---|---|
| Mutações sem sentido | Aproximadamente 39% |
| Mutações de deslocamento de quadro | Aproximadamente 25% |
| Mutações de splicing | Aproximadamente 13% |
| Mutação de sentido trocado | Cerca de 12% |
Mutações de readthrough (mutações de extensão C-terminal) representam cerca de 5%, resultando na produção de uma proteína PAX6 anormalmente alongada devido à conversão de um códon de parada em um códon de tradução 6). Mutações de extensão C-terminal frequentemente estão associadas a hipoplasia grave da íris e comprometimento visual severo 1)6).
As mutações genéticas são predominantemente do tipo PTC, mas também há relatos de mutações de sentido trocado 7). Quanto à utilidade dos testes genéticos, o sequenciamento de Sanger ou NGS detecta mutações em quase 85% dos casos de aniridia isolada. Além disso, MLPA ou CMA detectam deleções dentro do gene PAX6 ou em regiões cis-regulatórias em cerca de 15% dos casos 8).
Wang (2023) identificou uma nova mutação de deslocamento de quadro de leitura c.640_646del (p.R214Pfs*28) e relatou um caso com ausência completa da íris, displasia de fóvea, ectopia lentis e descolamento de retina 1).
Ratna et al. (2022) identificaram uma mutação de readthrough c.1268A>T (p.*423L) em uma família indiana. Os afetados apresentaram aniridia completa, nistagmo, displasia de fóvea, AAK, subluxação superior do cristalino, miopia alta e atrofia do nervo óptico, demonstrando um fenótipo grave devido à mutação de extensão C-terminal 6).
Na aniridia esporádica, deleções extensas que incluem o gene WT1 além de PAX6 causam a síndrome de WAGR. O risco de tumor de Wilms em pacientes com deleção de WT1 é de até 50% 1). Se houver suspeita de síndrome de WAGR, testes genéticos devem confirmar as deleções de PAX6 e WT1, permitindo a avaliação do risco de tumor de Wilms e o monitoramento do atraso no desenvolvimento 8). A avaliação da região WT1 por testes genéticos é essencial, e 30% dos casos esporádicos desenvolvem tumor de Wilms até os 5 anos de idade. Como o gene WT1 está localizado próximo ao PAX6, a deleção do braço curto do cromossomo 11 (deleção 11p13) que afeta ambos os genes resulta em aniridia associada a tumor de Wilms.
A síndrome de Gillespie é causada por mutações heterozigóticas dominantes negativas ou bialélicas no gene ITPR1 3). Até o momento, 37 casos com diagnóstico molecular confirmado foram relatados, sendo o resíduo Gly2554 um ponto crítico conhecido 3).
Com base nos critérios diagnósticos para aniridia (2020), o diagnóstico é confirmado pelos seguintes critérios7).
A. Sintomas
B. Achados de exames
C. Diagnósticos diferenciais
E. Teste genético: mutação patogênica no gene PAX6 ou deleção da região 11p13
Categorias diagnósticas7):
A classificação de gravidade para designação de doença rara é definida em quatro níveis7).
| Gravidade | Definição |
|---|---|
| Grau I | Acometimento unilateral, olho contralateral saudável |
| Grau II | Acometimento bilateral, melhor acuidade visual corrigida ≥ 0,3 |
| Grau III | Acometimento bilateral, melhor acuidade visual corrigida ≥ 0,1 e < 0,3 |
| Grau IV | Acometimento bilateral, melhor acuidade visual corrigida < 0,1 |
Mesmo nos graus I a III, se houver estreitamento do campo visual devido a glaucoma etc. (campo visual central residual ≤ 20° com o estímulo Goldmann I/4), a gravidade é elevada em um grau. Grau III ou superior é elegível para subsídio médico 7).
O diagnóstico clínico é fácil se a ausência ou hipoplasia da íris for confirmada por lâmpada de fenda. A avaliação do tecido iriano residual é realizada por gonioscopia ou microscopia ultrassônica de biomicroscopia. Também verifica-se a presença de anomalias de desenvolvimento do ângulo da câmara anterior.
As seguintes complicações oculares são avaliadas sistematicamente:
O objetivo mais importante na avaliação genética da aniridia é confirmar se a deleção do PAX6 se estende ao gene WT11). A análise de mutações e deleções nas regiões PAX6 e WT1 é realizada por sequenciamento completo do exoma ou MLPA1)2).
Na aniridia esporádica, a avaliação do risco de tumor de Wilms devido à deleção do gene WT1 está diretamente ligada ao prognóstico de vida1). Mesmo nos casos familiares, devido à diversidade fenotípica, recomenda-se o diagnóstico confirmatório por teste genético e aconselhamento genético.
Não existe tratamento curativo para a aniridia. O manejo centra-se nos cuidados de baixa visão para maximizar a visão residual e no tratamento individualizado de cada complicação8).
O transplante de córnea para opacidade do estroma corneano deve ser considerado com cautela8).
O transplante de córnea pode proporcionar melhora visual a curto prazo, mas a melhora é limitada devido a comorbidades como hipoplasia macular. A longo prazo, o prognóstico visual é ruim devido à progressão do glaucoma e à disfunção do enxerto.
Na deficiência de células-tronco epiteliais da córnea, considerar tratamento cirúrgico8).
A cirurgia de catarata deve ser considerada com base no grau de opacidade e fotofobia8).
A catarata se desenvolve em 50–85% dos pacientes até os 20 anos de idade. O planejamento cirúrgico é baseado na intensidade da opacidade e fotofobia. Relata-se melhora visual em 66–100% dos casos operados, mas os seguintes pontos requerem atenção:
Devido à fragilidade das zônulas de Zinn, a inserção de lente intraocular requer indicação cautelosa.
Hu et al. (2024) realizaram facoemulsificação assistida por iluminação retrógrada tipo candelabro em dois casos de aniridia congênita com AAK grave. Embora a visualização intraoperatória padrão fosse difícil devido à opacidade corneana, a iluminação posterior permitiu visualização clara do cristalino e da cápsula anterior, com melhora da acuidade visual corrigida para 20/200 e 20/1000 três semanas após a cirurgia 4).
O glaucoma deve ser tratado ativamente, pois impacta diretamente o prognóstico visual 8).
Após o início do glaucoma, o manejo segue um algoritmo de 5 etapas:
Terapia medicamentosa: Betabloqueadores, simpaticomiméticos e prostaglandinas (PG) são eficazes. Brimonidina (agonista alfa-adrenérgico) é contraindicada em menores de 2 anos devido ao risco de depressão do SNC. Se houver preocupação com lesão epitelial corneana, usar formulações sem conservantes.
Cirurgia reconstrutiva da via de drenagem (goniotomia/trabeculotomia): Recomendada como cirurgia inicial 16). Há relatos de goniotomia profilática. Pode ser ineficaz em casos onde a íris residual cobre a malha trabecular.
Cirurgia filtrante (trabeculectomia): Relatos limitados a poucos casos e médio-curto prazo. Tendência a piores resultados em olhos pediátricos, com fístula pós-operatória em cerca de 25% 13). Há relatos de glaucoma maligno pós-operatório.
Cirurgia de implante para glaucoma (tubo de drenagem): Dispositivos tipo Baerveldt e Ahmed estão disponíveis. Em olhos fácicos, recomenda-se inserção do tubo em direção tangencial, não central. Espera-se bom controle da pressão intraocular.
Ciclocoagulação: Último recurso. Relatos de que a ciclocriocoagulação levou à fístula em muitos casos. Devido à hipoplasia do corpo ciliar, o risco de fístula é maior do que em olhos saudáveis.
Devido à anomalia de desenvolvimento do ângulo, é necessária uma abordagem diferente da do glaucoma de ângulo aberto comum. Na primeira intervenção, opta-se pela cirurgia de reconstrução da via de drenagem, e a cirurgia de derivação com tubo é uma boa opção subsequente. A brimonidina é contraindicada em menores de 2 anos, e o uso de antimetabólitos pode agravar a AAK, exigindo julgamento cuidadoso 8).
Os cuidados para baixa visão devem ser introduzidos precocemente8).
A correção refrativa é fundamental, e a taxa de complicação de miopia é de 64% ou mais.
O tratamento da fotofobia é importante para preservar o desenvolvimento da função visual e a qualidade de vida8).
A maioria dos pacientes pode frequentar classes regulares, mas necessita de apoio como livros didáticos ampliados, tablets e estantes para leitura. A frequência em classes para visão subnormal ou o uso de consultas de aconselhamento infantil e educacional em escolas para cegos ou escolas especiais de apoio visual também são opções.
Desde abril de 2017, a doença foi designada como doença rara especificada, portanto, mesmo sem a obtenção do caderno de deficiência física, pacientes com gravidade grau III ou superior são elegíveis para subsídios médicos e fornecimento de órteses e próteses7). As órteses e próteses cobertas incluem óculos corretivos, óculos de proteção solar, lentes de contato (incluindo as com íris artificial), óculos para visão subnormal, bengala de segurança para deficientes visuais e olho artificial.
O PAX6 abrange 22 kb de DNA genômico, incluindo 14 éxons, e codifica 422 aminoácidos1). Possui dois domínios de ligação ao DNA (domínio pareado e homeodomínio pareado), e o domínio PST (rico em prolina, serina e treonina) na extremidade C-terminal funciona como ativador da transcrição.
O PAX6 regula a proliferação, diferenciação, migração e adesão celular, e seus alvos incluem o próprio PAX6, bem como genes que codificam cristalinas do cristalino e queratinas da córnea. A expressão continua na retina, cristalino e córnea de adultos. O gene PAX6 é um dos genes mestre de controle que governam a diferenciação de órgãos no período embrionário.
A maioria das mutações no PAX6 causa haploinsuficiência por meio da degradação do mRNA dependente de mutação sem sentido (NMD)1). Mutações que introduzem um códon de parada prematuro (PTC) (mutações sem sentido, mutações de deslocamento de quadro e a maioria das mutações de splicing) resultam no fenótipo típico de aniridia.
Por outro lado, se o PTC estiver localizado no último éxon ou dentro dos 50 bp terminais do penúltimo éxon, ele pode escapar do NMD, e uma proteína truncada pode ser traduzida, resultando em um fenótipo grave1).
Um caso raro foi relatado em que a mutação sem sentido c.282C>A (p.Cys94*) no PAX6 e a trissomia do 21 coexistiram no mesmo paciente. A mutação no PAX6 ocorreu de novo, resultando em aniridia bilateral completa, glaucoma congênito, AAK e hipoplasia da fóvea2).
Embora não haja uma correlação genótipo-fenótipo clara estabelecida, algumas tendências são conhecidas1).
Na série de gonioscopia de Grant e Walton, observou-se que, inicialmente, o estroma da íris se estende anteriormente sobre a malha trabecular, formando aderências semelhantes a sinéquias, que gradualmente se tornam em forma de lençol e, eventualmente, levam à oclusão angular14). Esse mecanismo é o principal fator no desenvolvimento do glaucoma. Patologicamente, a base é a ausência de músculo liso, com preservação da raiz da íris, e o desenvolvimento incompleto do ângulo.
A AAK é causada principalmente por deficiência de células-tronco do limbo (LSCD), mas também envolve diferenciação anormal do epitélio corneano, adesão anormal, infiltração de células conjuntivais e produção insuficiente de lágrimas. A deficiência de metaloproteinase de matriz 9 (MMP-9), regulada pelo PAX6, leva ao acúmulo de fibrina e infiltração de células inflamatórias, resultando em perda de transparência devido à desorganização das fibras de colágeno do estroma.
A AAK é classificada em 5 estágios. No Estágio I, há anormalidade apenas no epitélio periférico; no Estágio II, alterações epiteliais centrípetas (sem atingir o centro); no Estágio III, alterações epiteliais na córnea central e neovascularização superficial periférica; no Estágio IV, neovascularização superficial de toda a córnea; no Estágio V, anormalidades epiteliais em toda a córnea e cicatriz estromal profunda10).
Há uma relação entre o status da mutação do PAX6 e a progressão da AAK. Pacientes com mutações PTC ou de extensão C-terminal apresentam progressão da AAK dependente da idade, enquanto outros tipos de mutação podem resultar em ceratopatia não progressiva11).
A síndrome de Gillespie é causada por mutações no gene ITPR13). O ITPR1 é um membro da família de receptores IP3, formando canais de liberação de Ca²⁺ e localizando-se no retículo endoplasmático. Mutações dominantes negativas afetam a formação e manutenção do esfíncter da íris, resultando em disgenesia da íris específica ao redor da pupila e midríase fixa.
Na revisão da literatura sobre a síndrome de Gillespie por Ciaccio et al. (2024), a análise de 33 casos confirmados molecularmente mostrou que o desenvolvimento motor é atrasado, mas melhora ao longo do tempo; a deficiência intelectual não está presente em todos os casos, com 17% apresentando inteligência normal; e os sinais neurológicos são não progressivos3).
Com a disseminação da tecnologia de sequenciamento do exoma completo, novas mutações no PAX6 continuam sendo identificadas. Em 2018, o Human PAX6 Mutation Database registrava 491 mutações, e desde então cerca de 250 novas mutações foram relatadas1). Mutações em regiões não codificantes também estão sendo identificadas como causas de aniridia, o que pode esclarecer casos que não eram diagnosticados por exames convencionais9).
Para cirurgia de catarata em casos com AAK grave, a técnica de visualização com iluminação auxiliar tipo candelabro é útil4). Esse procedimento permite a facoemulsificação segura mesmo em pacientes com AAK de grau 3 a 4, resultando em melhora da acuidade visual pós-operatória.
Está se tornando evidente que diferentes tipos de mutações no PAX6 levam a padrões distintos de progressão da AAK. Com a redução dos custos dos testes genéticos, a predição da evolução clínica com base no tipo de mutação e a intervenção precoce estão se tornando opções viáveis.
Em casos de aniridia coexistente com trissomia do 21, foram relatados exemplos de evolução relativamente leve, apesar da presença de ambas as doenças2). Compreender o impacto no fenótipo quando múltiplos distúrbios genéticos coexistem no mesmo paciente pode trazer insights importantes para a medicina personalizada no futuro.
A aplicação de fármacos de leitura forçada (atalureno) para mutações do tipo PTC na aniridia está sendo investigada em nível de pesquisa básica8). Quanto à terapia gênica para PAX6, estudos básicos de reposição gênica com vetor AAV-PAX6 em modelos de camundongos com mutação Sey estão em andamento. Espera-se que futuramente haja ensaios clínicos.
Ensaios clínicos de transplante de folhas de células epiteliais da córnea derivadas de iPS estão sendo realizados no Japão e no exterior, atraindo atenção como nova terapia para AAK8). Quanto à íris artificial (como a CustomFlex Artificial Iris), há acúmulo de experiência de uso no exterior. Lentes de contato com íris artificial como órtese são cobertas pelo seguro de saúde.
O acúmulo de dados de grandes registros no Japão para compreender a situação real e melhorar a qualidade das evidências é uma questão importante para o futuro8). Espera-se a otimização da predição da progressão da AAK com base em mutações genéticas individuais e da intervenção precoce.