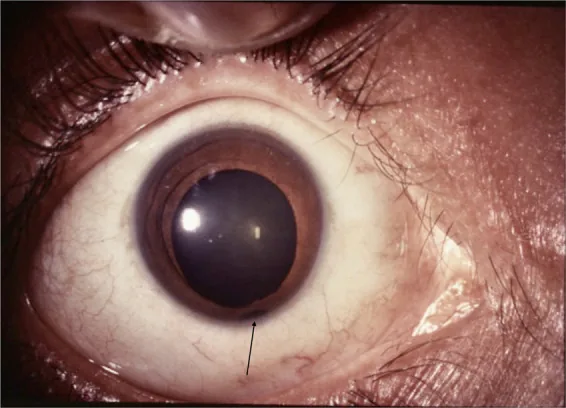
Íris
Pupila em buraco de fechadura: Tipicamente, o defeito localiza-se no lado inferonasal, deformando a pupila em forma de buraco de fechadura.
Localização inferotemporal: Pode ocorrer em posições atípicas.

Coloboma deriva do grego para “defeito” e é uma doença congênita caracterizada por falha no fechamento da fissura embrionária, resultando em defeitos teciduais em várias partes do olho. Pode ocorrer nas pálpebras, íris, cristalino, corpo ciliar, coroide, retina e nervo óptico. O defeito é tipicamente localizado na região inferonasal e frequentemente associado à microftalmia.
A prevalência é de 0,5 a 2,2 casos por 10.000 nascimentos. Nos EUA, é de aproximadamente 2,6 por 10.000 nascimentos 4), e na Europa, de 4 a 19 por 100.000 nascimentos 6). Representa cerca de 11% das causas de cegueira infantil, com taxa de diagnóstico genético inferior a 30% 6). A prevalência do coloboma palpebral é de 0,2 a 0,8 por 10.000 nascimentos. Corresponde a 0,07% das malformações oculares congênitas e a 3,2 a 11,2% das crianças com deficiência visual.
O coloboma pode ser típico ou atípico. O típico resulta da falha no fechamento da fissura embrionária e localiza-se na região inferonasal, enquanto o atípico ocorre em outras regiões e tem mecanismos de desenvolvimento diferentes.
Os códigos CID-10 são Q10.3 (pálpebra), Q13.0 (íris), Q12.2 (cristalino), H47.319 (nervo óptico) e Q14.8 (coroide e retina).
Existem formas esporádicas e hereditárias. Vários padrões de herança foram relatados, incluindo autossômica dominante, autossômica recessiva e ligada ao X. Múltiplos genes causadores foram identificados, como PAX2, CHD7 e FZD5, mas a taxa de diagnóstico genético é inferior a 30%6). Aconselhamento genético é recomendado se houver histórico familiar.
A acuidade visual varia amplamente, desde ausência de percepção luminosa até visão normal, dependendo da localização e extensão do defeito.
O coloboma apresenta achados característicos em cada parte do olho.
Íris
Pupila em buraco de fechadura: Tipicamente, o defeito localiza-se no lado inferonasal, deformando a pupila em forma de buraco de fechadura.
Localização inferotemporal: Pode ocorrer em posições atípicas.
Coroide e retina
Lesões amarelo-esbranquiçadas: áreas de perda de tecido bem delimitadas, redondas a em forma de leque, onde a esclera é visível.
Risco de descolamento de retina: incidência de 23 a 40%7). É necessário acompanhamento regular.
Nervo óptico e cristalino
Escavação aumentada do nervo óptico: pode ser unilateral ou bilateral, com gravidade variável.
Achatamento do equador do cristalino: ocorre devido à ausência das zônulas. Observado sob dilatação pupilar.
Pálpebras
Defeito na pálpebra superior medial: perda de tecido de espessura total.
Malformações sistêmicas associadas: Embora possa ocorrer isoladamente, frequentemente está associado a malformações sistêmicas.
O coloboma de corpo ciliar raramente ocorre isoladamente, sendo frequentemente contínuo a um grande coloboma coroideu.
A acuidade visual varia de ausência de percepção luminosa até normal. Colobomas isolados apenas da íris geralmente preservam a visão. Quando a mácula ou o nervo óptico estão envolvidos, a visão tende a ser ruim.
A principal causa do coloboma é o fechamento incompleto da fissura embrionária.
A fissura embrionária (fissura óptica) forma-se na 4ª semana de gestação e completa-se na 5ª semana. O fechamento inicia na 6ª semana e termina na 7ª semana. Se esse processo de fechamento for interrompido por alguma causa, ocorre o coloboma. A vitamina A também tem sido implicada.
Vários genes envolvidos na ocorrência de coloboma foram identificados.
| Gene | Doença/fenótipo associado |
|---|---|
| PAX2 | Síndrome do coloboma renal 5) |
| CHD7 | Síndrome de CHARGE |
| FZD5 | OC sintomático + microcórnea6) |
| TENM3 | MCOPS15 (microcórnea + atraso no desenvolvimento)8) |
| FAT1 | Coloboma + nefropatia9) |
| YAP1 | Relacionado a coloboma |
| ABCB6 | Relacionado a coloboma |
| SALL2 | Relacionado a coloboma |
O coloboma pode estar associado às seguintes síndromes sistêmicas.
É uma síndrome de múltiplas malformações causada por mutações no gene CHD7. O nome é um acrônimo para coloboma (C), cardiopatia (H), atresia de coanas (A), retardo de crescimento e desenvolvimento (R), hipoplasia genital (G) e anomalias de orelha (E), sendo diagnosticada pela combinação desses achados.
Testes genéticos abrangentes, como sequenciamento completo do exoma (WES), são realizados, mas a taxa de diagnóstico permanece abaixo de 30%6).
O coloboma requer diferenciação das seguintes doenças, dependendo da localização.
| Localização | Principais diagnósticos diferenciais |
|---|---|
| Pálpebra | Síndrome da brida amniótica, trauma |
| Íris | Aniridia, diálise traumática da íris |
| Nervo óptico | Síndrome do olho da manhã, hipoplasia do nervo óptico |
Não existe tratamento curativo para o coloboma; o tratamento é sintomático e focado no manejo de complicações, dependendo da localização.
Castilla-Martinez et al. (2024) combinaram cirurgia de catarata a laser femtossegundo (FLACS) com pupiloplastia e inseriram um ATC em um caso de coloboma da íris, cristalino e zônulas com catarata. A acuidade visual pós-operatória melhorou para logMAR 0,24).
No coloboma do nervo óptico, devido à displasia da lâmina cribrosa, a artéria e veia centrais da retina já se ramificam posteriormente à papila, e os vasos retinianos originam-se de múltiplos pontos na borda da papila. Inferiormente à papila, é frequente observar atrofia coriorretiniana devido ao fechamento incompleto da fissura embrionária.
Para o descolamento regmatogênico da retina, realiza-se vitrectomia. Técnicas cirúrgicas como retinopexia com cola de fibrina7) e fotocoagulação intraocular com tamponamento gasoso3) foram relatadas. No descolamento seroso, há casos de regressão espontânea, e a conduta é decidida individualmente.
O cálice óptico é formado a partir do neuroectoderma na 4ª semana de gestação. Na face ventral do cálice óptico, forma-se a fissura embrionária (fissura do cálice óptico), por onde passa a artéria hialoide. Essa fissura completa-se na 5ª semana e o fechamento inicia-se na 6ª semana. O fechamento começa próximo ao equador e progride em direção anterior (lado da íris) e posterior (lado do nervo óptico), completando-se na 7ª semana.
O processo de fechamento envolve a transição epitélio-mesenquimal (TEM). As células do epitélio neural da retina na borda da fissura embrionária degradam a membrana basal, adquirem fenótipo mesenquimal e se fundem. Quando esse processo é prejudicado, ocorre coloboma.
O gene FZD5 codifica um receptor da via de sinalização Wnt. Mutações de perda de função no FZD5 causam anormalidades na sinalização Wnt, resultando em falha no fechamento da fissura embrionária e microcórnea6).
As células da crista neural (CCN) também estão envolvidas na ocorrência do coloboma. As CCN diferenciam-se em tecido mesenquimal ao redor do cálice óptico e desempenham um papel importante no processo de fechamento da fissura embrionária2). A migração prejudicada das CCN leva a anormalidades no desenvolvimento da íris e da coroide.
Cortes-Gonzalez et al. (2024) relataram que uma mutação missense homozigótica em FZD5 (p.M160V) causa coloboma ocular sindrômico e microcórnea6). A herança é recessiva, e a análise funcional confirmou que a ativação dependente de ligante da via de sinalização Wnt está prejudicada. A taxa de diagnóstico genético do coloboma é inferior a 30%, e espera-se que a identificação de novos genes causadores contribua para melhorar o diagnóstico.
Zhou et al. (2022) relataram que mutações heterozigóticas compostas no gene TENM3 causam MCOPS15 (microcórnea, coloboma de íris e coroide, atraso global do desenvolvimento)8). TENM3 codifica uma proteína transmembrana envolvida na adesão celular e neurogênese.
Esmaeilzadeh et al. (2022) relataram que mutações no gene FAT1 foram identificadas em uma família iraniana com coloboma de íris e nefropatia9). FAT1 é um membro da superfamília das caderinas envolvido na polaridade celular e morfogênese tecidual.
Hu et al. (2024) relataram que a mutação frameshift c.76delG no PAX2 foi identificada em uma família com glomeruloesclerose segmentar e focal (GESF)5). Esse achado sugere que o espectro fenotípico da síndrome do coloboma renal é mais amplo do que se pensava anteriormente.
Jain et al. (2024) relataram um caso de descolamento de retina associado a coloboma tratado com retinopexia combinada com cola de fibrina7). A técnica envolve a aplicação de cola de fibrina ao redor dos rasgos retinianos na borda do coloboma para reforçar a adesão, resultando em acuidade visual final de 20/50.
Ratra et al. (2023) relataram sucesso no tratamento de um caso de coloboma coroideano atípico complicado por fístula escleral pós-traumática com vitrectomia, fotocoagulação endocular e tamponamento com gás3).
Scemla et al. (2021) relataram um caso de um homem de 19 anos com filtragem transescleral em um local de coloboma coroidal, resultando em hipotonia (4 mmHg)1). A microscopia ultrassônica biomicroscópica confirmou um defeito escleral. A recuperação espontânea ocorreu em 6 semanas, mantendo pressão intraocular de 11 mmHg e acuidade visual de 1,0.