Iris
Keyhole pupil: Typically, the defect is located inferonasally, causing the pupil to deform into a keyhole shape.
Inferotemporal location: It may also occur in atypical positions.

Coloboma derives from the Greek word meaning “defect” and is a congenital disease in which tissue defects occur in various parts of the eye due to incomplete closure of the embryonic fissure. It can occur in the eyelids, iris, lens, ciliary body, choroid, retina, and optic nerve. The defect is typically located inferonasally and is often accompanied by microphthalmia.
The prevalence is reported to be 0.5–2.2 per 10,000 births. In the United States, it is approximately 2.6 per 10,000 births 4), and in Europe, 4–19 per 100,000 births 6). It accounts for about 11% of childhood blindness, and the genetic diagnostic rate remains below 30% 6). The prevalence of eyelid coloboma is 0.2–0.8 per 10,000 births. It accounts for 0.07% of congenital eye malformations and 3.2–11.2% of visually impaired children.
Coloboma can be classified into typical and atypical types. Typical coloboma results from incomplete closure of the embryonic fissure and is located inferonasally, whereas atypical coloboma occurs in other locations and is thought to have a different developmental mechanism.
ICD-10 codes are Q10.3 (eyelid), Q13.0 (iris), Q12.2 (lens), H47.319 (optic nerve), and Q14.8 (choroid/retina).
Both sporadic and hereditary forms exist. Various inheritance patterns including autosomal dominant, autosomal recessive, and X-linked have been reported. Multiple causative genes such as PAX2, CHD7, and FZD5 have been identified, but the genetic diagnostic rate is less than 30% 6). Genetic counseling is recommended if there is a family history.
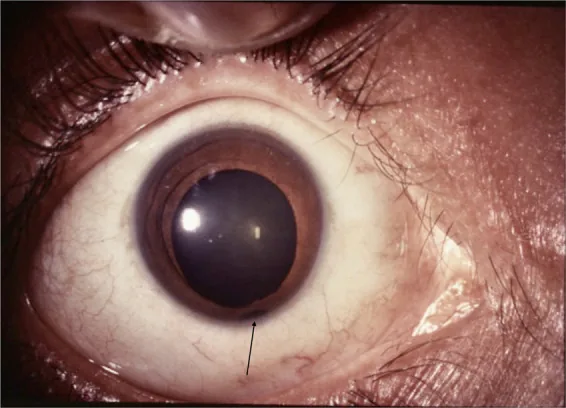
Visual acuity varies widely from no light perception to normal, depending on the location and extent of the defect.
Coloboma presents characteristic findings in each part of the eye.
Iris
Keyhole pupil: Typically, the defect is located inferonasally, causing the pupil to deform into a keyhole shape.
Inferotemporal location: It may also occur in atypical positions.
Choroid and Retina
Yellow-white lesion: A well-defined round to fan-shaped defect where the sclera is visible.
Risk of retinal detachment: Incidence 23–40%7). Regular follow-up is necessary.
Optic Nerve and Lens
Enlarged optic disc cupping: Varies in severity, from unilateral to bilateral.
Flattening of the lens equator: Occurs due to zonular defects. Observed under dilated pupils.
Eyelid
Defect in the upper eyelid medially: Full-thickness tissue defect.
Association with systemic malformations: May occur alone or with systemic anomalies.
Ciliary body coloboma rarely occurs alone and is often found contiguous with a large choroidal coloboma.
Visual acuity ranges from no light perception to normal. If the coloboma is limited to the iris, vision is often preserved. When the macula or optic nerve is involved, poor visual acuity is common.
The main cause of coloboma is failure of closure of the embryonic fissure.
The embryonic fissure (optic fissure) forms at 4 weeks of gestation and is completed by 5 weeks. Closure begins at 6 weeks and is completed by 7 weeks. If this closure process is disrupted for any reason, coloboma occurs. Involvement of vitamin A has also been suggested.
Multiple genes involved in the development of coloboma have been identified.
| Gene | Associated Disease/Phenotype |
|---|---|
| PAX2 | Renal coloboma syndrome5) |
| CHD7 | CHARGE syndrome |
| FZD5 | Syndromic OC + microcornea6) |
| TENM3 | MCOPS15 (microcornea + developmental delay) 8) |
| FAT1 | Coloboma + nephropathy 9) |
| YAP1 | Coloboma-related |
| ABCB6 | Coloboma-related |
| SALL2 | Coloboma-related |
Coloboma can be associated with the following systemic syndromes.
It is a multiple malformation syndrome caused by mutations in the CHD7 gene. The name is an acronym for coloboma (C), heart defects (H), choanal atresia (A), growth and developmental retardation (R), genital hypoplasia (G), and ear abnormalities (E). Diagnosis is based on a combination of these findings.
Comprehensive genetic testing such as whole exome sequencing (WES) is performed, but the diagnostic rate remains below 30% 6).
Coloboma requires differentiation from the following diseases depending on the site.
| Site | Main differential diagnoses |
|---|---|
| Eyelid | Amniotic band syndrome, trauma |
| Iris | Aniridia, traumatic iris dialysis |
| Optic nerve | Morning glory syndrome, optic nerve hypoplasia |
There is no curative treatment for coloboma; management focuses on symptomatic treatment and complication management according to the site.
Castilla-Martinez et al. (2024) performed femtosecond laser-assisted cataract surgery (FLACS) combined with pupilloplasty and placed a CTR in a case of iris, lens, and zonular coloboma with cataract. Postoperative visual acuity improved to logMAR 0.2 4).
In optic nerve coloboma, due to lamina cribrosa dysplasia, the central retinal artery and vein already branch posterior to the optic disc, and retinal vessels originate from multiple sites at the disc margin. Chorioretinal atrophy associated with incomplete closure of the embryonic fissure is often seen inferior to the disc.
Vitrectomy is performed for rhegmatogenous retinal detachment. Surgical techniques such as retinopexy with fibrin glue 7) and endophotocoagulation with gas tamponade 3) have been reported. For serous retinal detachment, spontaneous resolution may occur, and treatment is decided on a case-by-case basis.
The optic cup forms from the neuroectoderm at 4 weeks of gestation. On the ventral side of the optic cup, the embryonic fissure (optic cup fissure) develops, through which the hyaloid artery passes. This fissure is completed at 5 weeks, and closure begins at 6 weeks. Closure starts near the equator and proceeds anteriorly (toward the iris) and posteriorly (toward the optic nerve), completing at 7 weeks.
Epithelial-mesenchymal transition (EMT) is involved in the closure process. Neuroretinal epithelial cells at the edges of the embryonic fissure degrade the basement membrane, acquire mesenchymal characteristics, and fuse. Disruption of this process leads to coloboma.
The FZD5 gene encodes a receptor in the Wnt signaling pathway. Loss-of-function mutations in FZD5 cause abnormal Wnt signaling, leading to failure of embryonic fissure closure and microcornea 6).
Neural crest cells (NCCs) are also involved in coloboma development. NCCs differentiate into mesenchymal tissue around the optic cup and play an important role in the closure of the embryonic fissure 2). Impaired NCC migration leads to abnormalities in iris and choroid development.
Cortes-Gonzalez et al. (2024) reported that a homozygous missense mutation (p.M160V) in FZD5 causes syndromic ocular coloboma and microcornea 6). It shows an autosomal recessive inheritance pattern, and functional analysis confirmed impaired ligand-dependent activation of the Wnt signaling pathway. The genetic diagnostic rate for coloboma is less than 30%, and identification of novel causative genes is expected to improve diagnosis.
Zhou et al. (2022) reported that compound heterozygous mutations in the TENM3 gene cause MCOPS15 (microcornea, iris-choroidal coloboma, and global developmental delay) 8). TENM3 encodes a transmembrane protein involved in cell adhesion and neurodevelopment.
Esmaeilzadeh et al. (2022) reported that FAT1 gene mutations were identified in an Iranian family with iris coloboma and nephropathy 9). FAT1 is a member of the cadherin superfamily involved in cell polarity and tissue morphogenesis.
Hu et al. (2024) reported that a c.76delG frameshift mutation in PAX2 was identified in a family with focal segmental glomerulosclerosis (FSGS) 5). This finding suggests that the phenotypic spectrum of renal coloboma syndrome is broader than previously thought.
Jain et al. (2024) reported a case of coloboma-related retinal detachment treated with fibrin glue-assisted retinopexy 7). Fibrin glue was applied around the retinal tear at the coloboma margin to enhance adhesion, and final visual acuity improved to 20/50.
Ratra et al. (2023) reported successful treatment of a case of atypical choroidal coloboma complicated by post-traumatic scleral fistula using vitrectomy, endolaser photocoagulation, and gas tamponade 3).
Scemla et al. (2021) reported a case of a 19-year-old male with hypotony (4 mmHg) due to transscleral filtration at the site of choroidal coloboma 1). Scleral defect was confirmed by ultrasound biomicroscopy. Spontaneous recovery occurred within 6 weeks, maintaining intraocular pressure of 11 mmHg and visual acuity of 1.0.