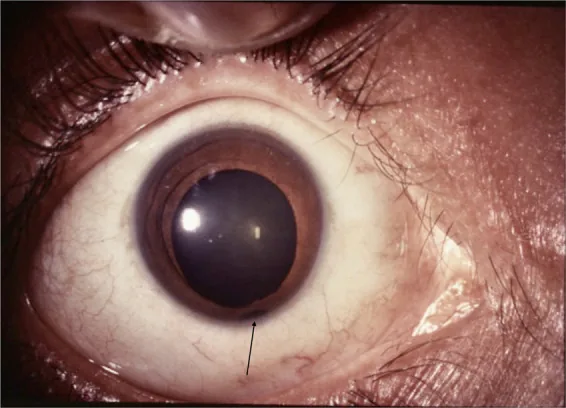
Iris
Pupila en ojo de cerradura: Típicamente, el defecto se localiza inferonasal, deformando la pupila en forma de ojo de cerradura.
Localización inferotemporal: También puede ocurrir en posiciones atípicas.

El coloboma deriva de la palabra griega que significa “defecto” y es una enfermedad congénita en la que se producen defectos tisulares en varias partes del ojo debido al cierre incompleto de la fisura embrionaria. Puede ocurrir en los párpados, iris, cristalino, cuerpo ciliar, coroides, retina y nervio óptico. El defecto se localiza típicamente en la región inferonasal y a menudo se acompaña de microftalmía.
La prevalencia se reporta entre 0.5 y 2.2 casos por cada 10,000 nacimientos. En Estados Unidos, es de aproximadamente 2.6 por cada 10,000 nacimientos 4), y en Europa, de 4 a 19 por cada 100,000 nacimientos 6). Representa aproximadamente el 11% de la ceguera infantil, y la tasa de diagnóstico genético es inferior al 30% 6). La prevalencia del coloboma palpebral es de 0.2 a 0.8 por cada 10,000 nacimientos. Representa el 0.07% de las malformaciones oculares congénitas y entre el 3.2% y el 11.2% de los niños con discapacidad visual.
El coloboma puede ser típico o atípico. El coloboma típico se debe al cierre incompleto de la fisura embrionaria y se localiza en la región inferonasal, mientras que el atípico ocurre en otras ubicaciones y se cree que tiene un mecanismo de desarrollo diferente.
Los códigos CIE-10 son Q10.3 (párpado), Q13.0 (iris), Q12.2 (cristalino), H47.319 (nervio óptico) y Q14.8 (coroides/retina).
Existen formas esporádicas y hereditarias. Se han reportado diversos patrones de herencia, incluyendo autosómico dominante, autosómico recesivo y ligado al cromosoma X. Se han identificado múltiples genes causantes como PAX2, CHD7 y FZD5, pero la tasa de diagnóstico genético es inferior al 30% 6). Se recomienda asesoramiento genético si hay antecedentes familiares.
La agudeza visual varía ampliamente desde ausencia de percepción de luz hasta normal, dependiendo de la ubicación y extensión del defecto.
El coloboma presenta hallazgos característicos en cada parte del ojo.
Iris
Pupila en ojo de cerradura: Típicamente, el defecto se localiza inferonasal, deformando la pupila en forma de ojo de cerradura.
Localización inferotemporal: También puede ocurrir en posiciones atípicas.
Coroides y Retina
Lesión amarillenta: Defecto bien delimitado, redondo a forma de abanico, donde se trasluce la esclerótica.
Riesgo de desprendimiento de retina: Incidencia del 23–40%7). Se requiere seguimiento regular.
Nervio Óptico y Cristalino
Agrandamiento de la excavación del disco óptico: Varía en severidad, desde unilateral a bilateral.
Aplanamiento del ecuador del cristalino: Ocurre debido a defectos zonulares. Se observa bajo dilatación pupilar.
Párpado
Defecto en el párpado superior medial: Defecto de espesor total.
Asociación con malformaciones sistémicas: Puede ocurrir solo o con anomalías sistémicas.
El coloboma del cuerpo ciliar rara vez ocurre solo y a menudo se encuentra contiguo a un coloboma coroideo grande.
La agudeza visual varía desde ausencia de percepción de luz hasta normal. Si el coloboma se limita al iris, la visión suele conservarse. Cuando afecta la mácula o el nervio óptico, es frecuente una mala agudeza visual.
La causa principal del coloboma es el cierre incompleto de la fisura embrionaria.
La fisura embrionaria (fisura óptica) se forma en la semana 4 de gestación y se completa en la semana 5. El cierre comienza en la semana 6 y finaliza en la semana 7. Si este proceso de cierre se ve alterado por alguna razón, se produce el coloboma. También se ha señalado la participación de la vitamina A.
Se han identificado múltiples genes implicados en el desarrollo del coloboma.
| Gen | Enfermedad/Fenotipo asociado |
|---|---|
| PAX2 | Síndrome de coloboma renal5) |
| CHD7 | Síndrome CHARGE |
| FZD5 | OC sindrómico + microcórnea6) |
| TENM3 | MCOPS15 (microcórnea + retraso del desarrollo) 8) |
| FAT1 | Coloboma + nefropatía 9) |
| YAP1 | Relacionado con coloboma |
| ABCB6 | Relacionado con coloboma |
| SALL2 | Relacionado con coloboma |
El coloboma puede asociarse con los siguientes síndromes sistémicos.
Es un síndrome de malformaciones múltiples causado por mutaciones en el gen CHD7. El nombre es un acrónimo de coloboma (C), defectos cardíacos (H), atresia de coanas (A), retraso del crecimiento y desarrollo (R), hipoplasia genital (G) y anomalías del oído (E). Se diagnostica por la combinación de estos hallazgos.
Se realizan pruebas genéticas integrales como la secuenciación del exoma completo (WES), pero la tasa de diagnóstico sigue siendo inferior al 30% 6).
El coloboma requiere diferenciación de las siguientes enfermedades según la ubicación.
| Ubicación | Principales diagnósticos diferenciales |
|---|---|
| Párpado | Síndrome de bandas amnióticas, traumatismo |
| Iris | Aniridia, diálisis traumática del iris |
| Nervio óptico | Síndrome de morning glory, hipoplasia del nervio óptico |
No existe un tratamiento curativo para el coloboma; el manejo se centra en el tratamiento sintomático y el manejo de complicaciones según la ubicación.
Castilla-Martinez et al. (2024) realizaron cirugía de cataratas asistida por láser de femtosegundo (FLACS) combinada con pupilaplastia y colocaron un CTR en un caso de coloboma de iris, cristalino y zónula con catarata. La agudeza visual postoperatoria mejoró a logMAR 0.2 4).
En el coloboma del nervio óptico, debido a la displasia de la lámina cribosa, la arteria y vena centrales de la retina ya se ramifican posterior a la papila, y los vasos retinianos se originan en múltiples sitios en el margen de la papila. A menudo se observa atrofia coriorretiniana inferior a la papila asociada con el cierre incompleto de la fisura embrionaria.
Se realiza vitrectomía para el desprendimiento de retina regmatógeno. Se han reportado técnicas quirúrgicas como la retinopexia con pegamento de fibrina 7) y la fotocoagulación endocular con taponamiento con gas 3). En el desprendimiento de retina seroso, puede ocurrir resolución espontánea, y el tratamiento se decide de forma individualizada.
La copa óptica se forma a partir del neuroectodermo en la cuarta semana de gestación. En el lado ventral de la copa óptica se desarrolla la fisura embrionaria (fisura de la copa óptica), a través de la cual pasa la arteria hialoidea. Esta fisura se completa en la quinta semana y el cierre comienza en la sexta semana. El cierre comienza cerca del ecuador y progresa hacia adelante (hacia el iris) y hacia atrás (hacia el nervio óptico), completándose en la séptima semana.
El proceso de cierre involucra la transición epitelio-mesenquimatosa (TEM). Las células del epitelio neuroretiniano en los bordes de la fisura embrionaria degradan la membrana basal, adquieren características mesenquimatosas y se fusionan. La alteración de este proceso conduce al coloboma.
El gen FZD5 codifica un receptor en la vía de señalización Wnt. Las mutaciones de pérdida de función en FZD5 causan señalización Wnt anormal, lo que lleva a un cierre incompleto de la fisura embrionaria y microcórnea 6).
Las células de la cresta neural (CCN) también están involucradas en el desarrollo del coloboma. Las CCN se diferencian en tejido mesenquimatoso alrededor de la copa óptica y desempeñan un papel importante en el cierre de la fisura embrionaria 2). La alteración de la migración de las CCN conduce a anomalías en el desarrollo del iris y la coroides.
Cortes-Gonzalez et al. (2024) informaron que una mutación missense homocigota (p.M160V) en FZD5 causa coloboma ocular sindrómico y microcórnea 6). Presenta un patrón de herencia recesivo, y el análisis funcional confirmó una alteración en la activación dependiente de ligando de la vía de señalización Wnt. La tasa de diagnóstico genético del coloboma es inferior al 30%, y se espera que la identificación de nuevos genes causantes contribuya a mejorar el diagnóstico.
Zhou et al. (2022) informaron que mutaciones heterocigotas compuestas en el gen TENM3 causan MCOPS15 (microcórnea, coloboma iridocoroideo y retraso global del desarrollo) 8). TENM3 codifica una proteína transmembrana involucrada en la adhesión celular y el neurodesarrollo.
Esmaeilzadeh et al. (2022) informaron que mutaciones en el gen FAT1 se identificaron en una familia iraní con coloboma de iris y nefropatía 9). FAT1 es un miembro de la superfamilia de cadherinas involucrado en la polaridad celular y la morfogénesis tisular.
Hu et al. (2024) informaron que una mutación de cambio de marco c.76delG en PAX2 se identificó en una familia con glomeruloesclerosis focal y segmentaria (GEFS) 5). Este hallazgo sugiere que el espectro fenotípico del síndrome de coloboma renal es más amplio de lo que se pensaba anteriormente.
Jain et al. (2024) informaron un caso de desprendimiento de retina relacionado con coloboma tratado con retinopexia asistida con pegamento de fibrina 7). Se aplicó pegamento de fibrina alrededor del desgarro retiniano en el margen del coloboma para mejorar la adhesión, y la agudeza visual final mejoró a 20/50.
Ratra et al. (2023) informaron el tratamiento exitoso de un caso de coloboma coroideo atípico complicado con fístula escleral postraumática mediante vitrectomía, fotocoagulación endocular y taponamiento con gas 3).
Scemla et al. (2021) informaron un caso de un varón de 19 años con hipotonía (4 mmHg) debido a filtración transescleral en el sitio de un coloboma coroideo 1). Se confirmó un defecto escleral mediante microscopía ultrasónica biomicroscópica. Se produjo una recuperación espontánea en 6 semanas, manteniendo una presión intraocular de 11 mmHg y una agudeza visual de 1.0.