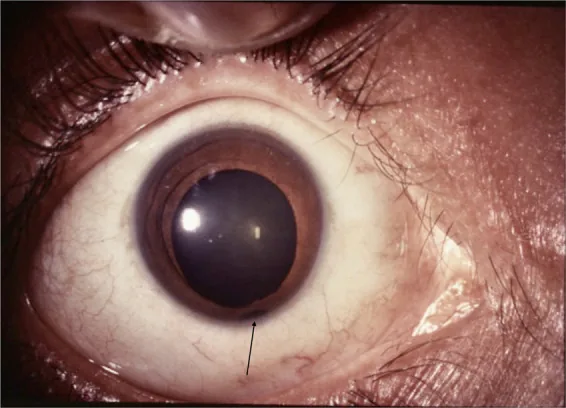
Iris
Schlüssellochpupille : Typischerweise liegt der Defekt unten-nasal, die Pupille ist schlüssellochförmig deformiert.
Unten-temporal : Kann auch an atypischen Stellen auftreten.

Das Kolobom (coloboma) leitet sich vom griechischen Wort für „Defekt“ ab und ist eine angeborene Erkrankung, bei der es aufgrund eines unvollständigen Verschlusses der embryonalen Spalte zu Gewebedefekten in verschiedenen Teilen des Auges kommt. Es kann an den Augenlidern, der Iris, der Linse, dem Ziliarkörper, der Aderhaut, der Netzhaut und dem Sehnerv auftreten. Der Defekt liegt typischerweise inferonasal und ist häufig mit einer Mikrophthalmie verbunden.
Die Prävalenz wird auf 0,5 bis 2,2 Fälle pro 10.000 Geburten geschätzt. In den USA liegt sie bei etwa 2,6 Fällen pro 10.000 Geburten 4), in Europa bei 4 bis 19 Fällen pro 100.000 Geburten 6). Sie macht etwa 11 % der kindlichen Erblindungen aus, und die genetische Diagnoserate beträgt weniger als 30 % 6). Die Prävalenz des Lidkoloboms liegt bei 0,2 bis 0,8 Fällen pro 10.000 Geburten. Es macht 0,07 % der angeborenen Augenfehlbildungen und 3,2 bis 11,2 % bei sehbehinderten Kindern aus.
Es gibt typische und atypische Kolobome. Typische Kolobome entstehen durch einen unvollständigen Verschluss der embryonalen Spalte und liegen inferonasal, während atypische Kolobome an anderen Stellen auftreten und unterschiedliche Entwicklungsmechanismen vermuten lassen.
Die ICD-10-Codes sind Q10.3 (Lid), Q13.0 (Iris), Q12.2 (Linse), H47.319 (Sehnerv) und Q14.8 (Aderhaut/Netzhaut).
Es gibt sowohl sporadische als auch erbliche Formen. Es wurden verschiedene Vererbungsmuster berichtet, darunter autosomal-dominant, autosomal-rezessiv und X-chromosomal. Mehrere ursächliche Gene wie PAX2, CHD7 und FZD5 wurden identifiziert, aber die genetische Diagnoserate liegt unter 30 % 6). Bei positiver Familienanamnese wird eine genetische Beratung empfohlen.
Die Sehschärfe variiert stark, von fehlender Lichtwahrnehmung bis normal, abhängig von Ort und Ausmaß des Defekts.
Das Kolobom zeigt in jedem Teil des Auges charakteristische Befunde.
Iris
Schlüssellochpupille : Typischerweise liegt der Defekt unten-nasal, die Pupille ist schlüssellochförmig deformiert.
Unten-temporal : Kann auch an atypischen Stellen auftreten.
Aderhaut und Netzhaut
Gelb-weiße Herde : Scharf begrenzter, runder bis fächerförmiger Defekt, durch den die Sklera durchscheint.
Netzhautablösungsrisiko : Inzidenz 23–40 %7). Regelmäßige Nachkontrollen erforderlich.
Sehnerv und Linse
Vergrößerte Sehnervenexkavation : Ein- bis beidseitig, unterschiedlichen Ausmaßes.
Abflachung des Linsenäquators : Aufgrund eines Zonuladefekts. Unter Mydriasis beobachtbar.
Lid
Innerer Defekt des Oberlids : Vollschichtiger Gewebedefekt.
Assoziation mit systemischen Fehlbildungen : Manchmal isoliert, kann aber mit systemischen Fehlbildungen einhergehen.
Ein isoliertes Ziliarkörperkolobom ist selten; es wird häufig in Kontinuität mit einem großen Aderhautkolobom gefunden.
Die Sehschärfe reicht von keiner Lichtwahrnehmung bis normal. Ist das Kolobom auf die Iris beschränkt, bleibt das Sehvermögen oft erhalten. Bei Beteiligung der Makula oder des Sehnervs ist die Sehschärfe tendenziell schlecht.
Die Hauptursache für ein Kolobom ist der unvollständige Verschluss der embryonalen Spalte.
Die embryonale Spalte (Augenbecherspalte) bildet sich in der 4. Schwangerschaftswoche und ist in der 5. Woche vollständig. Der Verschluss beginnt in der 6. Woche und ist in der 7. Woche abgeschlossen. Wird dieser Verschlussprozess aus irgendeinem Grund gestört, entsteht ein Kolobom. Auch eine Beteiligung von Vitamin A wird vermutet.
Mehrere Gene, die an der Entstehung von Kolobomen beteiligt sind, wurden identifiziert.
| Gen | Assoziierte Erkrankung/Phänotyp |
|---|---|
| PAX2 | Nieren-Kolobom-Syndrom5) |
| CHD7 | CHARGE-Syndrom |
| FZD5 | Symptomatisches okuläres Kolobom + Mikrokornea6) |
| TENM3 | MCOPS15 (Mikrokornea + Entwicklungsverzögerung) 8) |
| FAT1 | Kolobom + Nephropathie 9) |
| YAP1 | Kolobom-assoziiert |
| ABCB6 | Kolobom-assoziiert |
| SALL2 | Kolobom-assoziiert |
Ein Kolobom kann mit folgenden systemischen Syndromen einhergehen.
Es handelt sich um ein multiples Fehlbildungssyndrom, das durch eine Mutation im CHD7-Gen verursacht wird. Der Name setzt sich aus den Anfangsbuchstaben von Kolobom (C), Herzfehler (H), Choanalatresie (A), Wachstums- und Entwicklungsverzögerung (R), Genitalhypoplasie (G) und Ohranomalien (E) zusammen. Die Diagnose basiert auf einer Kombination dieser Befunde.
Umfassende Gentests wie die Exomsequenzierung (WES) werden durchgeführt, aber die Diagnoserate liegt unter 30 %6).
Ein Kolobom erfordert je nach Lokalisation die Abgrenzung zu folgenden Erkrankungen.
| Lokalisation | Wichtigste Differenzialdiagnosen |
|---|---|
| Augenlid | Amnionbandsyndrom, Trauma |
| Iris | Aniridie, traumatische Iridodialyse |
| Sehnerv | Morning-Glory-Syndrom, Optikushypoplasie |
Es gibt keine kausale Therapie für Kolobome; die Behandlung besteht aus symptomatischen Maßnahmen und Komplikationsmanagement je nach Lokalisation.
Castilla-Martinez et al. (2024) führten bei einem Fall von Iris-, Linsen- und Zonulakolobom mit Katarakt eine Femtosekundenlaser-Kataraktoperation (FLACS) in Kombination mit einer Pupilloplastik und CTR-Einlage durch. Die postoperative Sehschärfe verbesserte sich auf logMAR 0,24).
Beim Sehnervenkolobom verzweigen sich aufgrund einer Hypoplasie der Lamina cribrosa die zentrale Netzhautarterie und -vene bereits hinter der Papille, und die Netzhautgefäße entspringen an mehreren Stellen am Papillenrand. Unterhalb der Papille findet sich häufig eine Aderhaut-Netzhaut-Atrophie infolge eines unvollständigen Verschlusses der embryonalen Spalte.
Bei rhegmatogener Netzhautablösung wird eine Vitrektomie durchgeführt. Chirurgische Techniken wie die Netzhautwiederanlage mit Fibrinkleber 7) und Endolaserkoagulation mit Gastamponade 3) wurden beschrieben. Bei seröser Netzhautablösung kann eine spontane Rückbildung auftreten, und die Behandlungsstrategie wird individuell festgelegt.
Der Augenbecher bildet sich in der 4. Embryonalwoche aus dem Neuroektoderm. Auf der ventralen Seite des Augenbechers entsteht eine embryonale Spalte (Augenbecherspalte), durch die die Arteria hyaloidea verläuft. Diese Spalte ist in der 5. Woche vollständig ausgebildet und beginnt sich ab der 6. Woche zu schließen. Der Verschluss beginnt in Äquatornähe und schreitet nach vorne (zur Iris hin) und nach hinten (zum Sehnerv hin) fort; er ist in der 7. Woche abgeschlossen.
Am Verschlussprozess ist die epithelial-mesenchymale Transition (EMT) beteiligt. Die Neuroretina-Epithelzellen am Rand der embryonalen Spalte bauen die Basalmembran ab, erwerben einen mesenchymalen Phänotyp und fusionieren. Eine Störung dieses Prozesses führt zur Entstehung eines Koloboms.
Das FZD5-Gen kodiert für einen Rezeptor des Wnt-Signalwegs. Funktionsverlustmutationen von FZD5 verursachen eine Anomalie der Wnt-Signalgebung, die zu einem unvollständigen Verschluss der embryonalen Spalte und einer Mikrokornea führt 6).
Auch Neuralleistenzellen (NCC) sind an der Entstehung von Kolobomen beteiligt. NCC differenzieren sich zu mesenchymalen Gewebe um den Augenbecher und spielen eine wichtige Rolle beim Verschluss der embryonalen Spalte 2). Eine Störung der NCC-Migration führt zu Entwicklungsanomalien von Iris und Aderhaut.
Cortes-Gonzalez et al. (2024) berichteten, dass eine homozygote Missense-Mutation in FZD5 (p.M160V) symptomatisches okuläres Kolobom und Mikrokornea verursacht 6). Es zeigt einen rezessiven Erbgang, und die funktionelle Analyse bestätigte eine Beeinträchtigung der ligandenabhängigen Aktivierung des Wnt-Signalwegs. Die genetische Diagnoserate für Kolobome liegt unter 30 %, und die Identifizierung neuer ursächlicher Gene wird voraussichtlich die Diagnose verbessern.
Zhou et al. (2022) berichteten, dass compound-heterozygote Mutationen im TENM3-Gen MCOPS15 (Mikrokornea, Iris-Choroidea-Kolobom, globale Entwicklungsverzögerung) verursachen 8). TENM3 kodiert ein Transmembranprotein, das an Zelladhäsion und Neurogenese beteiligt ist.
Esmaeilzadeh et al. (2022) berichteten, dass Mutationen im FAT1-Gen in einer iranischen Familie mit Iris-Kolobom und Nephropathie identifiziert wurden 9). FAT1 ist ein Mitglied der Cadherin-Superfamilie, das an Zellpolarität und Gewebemorphogenese beteiligt ist.
Hu et al. (2024) berichteten, dass eine Frameshift-Mutation c.76delG in PAX2 in einer Familie mit fokal-segmentaler Glomerulosklerose (FSGS) identifiziert wurde 5). Dieser Befund deutet darauf hin, dass das phänotypische Spektrum des renalen Kolobom-Syndroms breiter ist als bisher angenommen.
Jain et al. (2024) berichteten über einen Fall von kolobomassoziierter Netzhautablösung, der mit einer Retinopexie unter Verwendung von Fibrinkleber behandelt wurde 7). Die Technik besteht darin, Fibrinkleber um den Netzhautriss am Rand des Koloboms aufzutragen, um die Adhäsion zu verstärken; die endgültige Sehschärfe verbesserte sich auf 20/50.
Ratra et al. (2023) berichteten über einen Fall eines atypischen Aderhautkoloboms, das durch eine posttraumatische Skleralfistel kompliziert wurde und erfolgreich mit Vitrektomie, endokularer Photokoagulation und Gastamponade behandelt wurde 3).
Scemla et al. (2021) berichteten über einen Fall eines 19-jährigen Mannes mit transskleraler Filtration an der Stelle eines Aderhautkoloboms, die zu einer Hypotonie (4 mmHg) führte 1). Die Ultraschallbiomikroskopie bestätigte einen Skleradefekt. Die spontane Erholung erfolgte innerhalb von 6 Wochen, der Augeninnendruck betrug 11 mmHg und die Sehschärfe blieb bei 1,0.