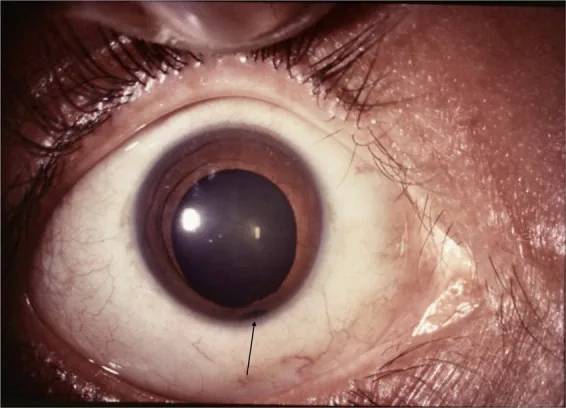
Das Uveakolobom (Coloboma uveae) ist eine angeborene Augenfehlbildung, die durch einen unvollständigen Verschluss der embryonalen Spalte verursacht wird. Der Defekt tritt an der Sehnervenpapille, der Aderhaut, dem Ziliarkörper und der Iris entsprechend der embryonalen Spalte auf. Tritt er an der Unterseite des Augenbechers auf, spricht man von einem typischen Kolobom, an anderen Stellen (z. B. Makulakolobom) von einem atypischen Kolobom.
Das typische Iriskolobom (Coloboma iridis) entsteht durch einen unvollständigen Verschluss der Augenbecherspalte. Der Augenbecher umschließt das Gewebe, das den Augeninhalt bilden soll, von oben und schließt sich schließlich unten. Daher liegt das typische Iriskolobom unten (etwas zur unteren Schläfenseite hin) und kann mit einem Ziliarkörperkolobom, Aderhautkolobom und Sehnervenkolobom einhergehen. Das Iriskolobom kann isoliert auftreten, ist aber häufig mit anderen Uveakolobomen kombiniert.
Es gibt derzeit keine Behandlung, die das Iriskolobom selbst „heilen“ kann. Die Behandlungsziele sind die symptomatische Behandlung der Photophobie, das Management und die Prävention von Komplikationen sowie die Optimierung der Sehfunktion.
Kann als Symptom von Multiorganfehlbildungen wie dem CHARGE-Syndrom (CHD7-Mutation) entdeckt werden.
QIst das Iriskolobom vererbbar?
A
Die Vererbungsmuster sind vielfältig: Es kann sporadisch auftreten oder autosomal-dominant (AD) oder autosomal-rezessiv (AR) bei mehreren Familienmitgliedern vorkommen. Es kann auch als Symptom eines Multiorgan-Fehlbildungssyndroms wie dem CHARGE-Syndrom (CHD7-Mutation, AD) auftreten. Bei positiver Familienanamnese oder Verdacht auf ein Syndrom wird eine genetische Beratung empfohlen.
Lingam G, et al. Ocular coloboma-a comprehensive review for the clinician. Eye (Lond). 2021. Figure 2. PMCID: PMC8302742. License: CC BY.
Vorderabschnittsfoto, das ein Iriskolobom mit einer schlüssellochförmigen Einkerbung im unteren Pupillenbereich deutlich zeigt. Es stellt einen repräsentativen klinischen Befund des Iriskoloboms dar und eignet sich zur Beschreibung der Hauptsymptome und klinischen Befunde.
Sehschärfe: Bei isoliertem Iriskolobom ist die Sehschärfe oft gut. Reicht der Defekt bis zur Makula, ist die Sehschärfe schlecht.
Photophobie (Lichtempfindlichkeit): Der durch den Irisdefekt verursachte Mangel an Lichtregulation führt zu Photophobie. Dies kann im Alltag eine starke Beeinträchtigung darstellen.
Nystagmus: Bei beidseitigem Befall kann ein Nystagmus auftreten. Besonders ausgeprägt ist er bei schlechter Sehschärfe.
Der Defekt liegt inferotemporal, ist am Pupillenrand am breitesten und verjüngt sich zur Iriswurzel hin (schlüssellochförmige Pupille: Keyhole-Pupille). Häufig begleitet von Mikrophthalmie, Katarakt oder Aderhautkolobom.
Aussehen: Schlüssellochförmige Pupillendeformität. Am Pupillenrand am breitesten, zur Wurzel hin schmaler werdend.
Hauptsymptome: Photophobie durch Störung der Lichtregulation. Bei isoliertem Defekt ist die Sehschärfe oft gut.
Ziliarkörperkolobom
Lokalisation: Häufig durchgehend mit einem Irisdefekt.
Merkmale: Isoliertes Auftreten selten. Häufig durchgehend mit einem großen Aderhautkolobom. Defekt von Epithel und Muskel.
Komplikationen: Schwächung der Zonula Zinnii. Verminderte Linsenunterstützung.
Aderhautkolobom
Befund: Als scharf begrenzte, eingesunkene gelb-weiße Läsion im unteren Fundusbereich zu beobachten.
Komplikationen: Netzhautrissbildung am Defektrand → hohes Risiko einer Netzhautablösung. Ausdehnung auf die Makula führt zu schlechter Sehprognose.
Sehnervenkolobom
Befund: Vergrößerung und Exkavation der Papille. Der Defekt breitet sich um die Papille aus.
Komplikationen: Gesichtsfeldausfälle und Optikusatrophie. Große Auswirkung auf das Sehvermögen.
Nach der phänotypischen Klassifikation von Hornby et al. ist die Ausdehnung des Koloboms auf den hinteren Augenabschnitt der Hauptfaktor für die Sehprognose: Je weiter der Defekt auf die Makula übergreift, desto schlechter ist die Prognose8).
QWelche Auswirkungen hat ein Irisdefekt auf das Sehvermögen?
A
Ein isolierter Irisdefekt geht oft mit gutem Sehvermögen einher. Wenn sich jedoch ein Aderhautkolobom auf die Makula ausdehnt, nimmt die Sehkraft stark ab und die Prognose ist schlecht. Bei beidseitigem Auftreten kann ein Nystagmus auftreten und eine Amblyopie kann bestehen. Regelmäßige augenärztliche Kontrollen des Sehvermögens und des Fundus sind wichtig.
Um die 6. bis 7. Schwangerschaftswoche herum erfolgt der Verschluss der embryonalen Spalte (Augenbecherspalte / Aderhautspalte). Der Verschluss beginnt am unteren Äquator und schreitet in beide Richtungen zum hinteren Pol und zur Peripherie fort. Daher neigen die hintere Aderhaut und der untere Teil der Iris dazu, als Kolobom zurückzubleiben. Wenn der Verschluss unvollständig ist, bilden sich Kolobome in der unteren Uvea (Iris, Ziliarkörper, Aderhaut) und im Sehnerv.
Hinsichtlich Umweltfaktoren wurde in Tierversuchen die Beteiligung teratogener Substanzen berichtet, aber die etablierten Risikofaktoren beim Menschen sind begrenzt2).
Es handelt sich um ein Multiorgan-Fehlbildungssyndrom, das durch eine Mutation des CHD7-Gens verursacht wird. Der Name leitet sich vom Akronym seiner Hauptbefunde ab: Kolobom, Herzfehler, Choanalatresie, Wachstums-/Entwicklungsverzögerung, Genitalanomalien, Ohranomalien. Die Vererbung ist autosomal-dominant, und das Kolobom ist oft der Ausgangspunkt für die Diagnose. Für die systemische Behandlung ist eine multidisziplinäre Zusammenarbeit erforderlich.
Spaltlampenmikroskopie: Die Untersuchung des vorderen Augenabschnitts zeigt einen unteren Irisdefekt (schlüssellochförmige Pupille). Dies ist der Hauptbefund für die definitive Diagnose.
Fundusuntersuchung in Mydriasis: Bestätigung eines Aderhautkoloboms und eines Sehnervenkoloboms. Beurteilung einer Beteiligung der Makula.
Die Abgrenzung zur Aniridie ist klinisch wichtig. Die Diagnosekriterien für Aniridie (Jpn J Ophthalmol 2020) führen das Iriskolobom ausdrücklich als Differenzialdiagnose auf4).
PAX6-Mutation. Iris nahezu vollständig fehlend (>2/3). Häufig begleitende Keratopathie und Makulahypoplasie4). Das Risiko eines Wilms-Tumors (WAGR-Syndrom) ist spezifisch für die Aniridie und tritt beim Iriskolobom in der Regel nicht auf.
Membranöse Struktur verbleibt im Pupillarbereich. Das Irisstroma ist nicht defekt
QWann und wie wird ein Iriskolobom entdeckt?
A
Meist wird es bei der Geburt oder im Säuglingsalter bei der Untersuchung des vorderen Augenabschnitts als schlüssellochförmige Pupille entdeckt. Die Diagnose wird mit der Spaltlampe bestätigt, und eine Fundusuntersuchung in Mydriasis überprüft auf Komplikationen der Aderhaut oder des Sehnervs. Bei Verdacht auf CHARGE-Syndrom erfolgt eine allgemeine Untersuchung von Herz, Ohren und Nase. Es kann auch zufällig bei Schuluntersuchungen oder Vorsorgeuntersuchungen entdeckt werden.
Es gibt keine kurative Behandlung des Iriskoloboms selbst. Die symptomatische Behandlung der Photophobie, des Hauptsymptoms, ist der Ausgangspunkt der Therapie.
Lichtschutzbrille: Grundlegendste Maßnahme, um die durch das Iriskolobom verursachte Lichtregulationsstörung auszugleichen und die Photophobie zu reduzieren. Der Grad der Abdunklung und der Farbfilter werden je nach Symptomatik gewählt.
Farbige Kontaktlinsen mit Iris (prothetische CL): Ermöglichen sowohl eine kosmetische Verbesserung als auch eine Reduktion der Photophobie. Sie ergänzen das Iriskolobom optisch.
Refraktionskorrektur: Korrektur von Refraktionsfehlern mit Brille oder normalen Kontaktlinsen.
Amblyopieprävention: Bei Kindern sind eine angemessene Refraktionskorrektur und die Okklusion des gesunden Auges wichtig zur Vorbeugung von Amblyopie und Strabismus.
Besonderheiten: Die Operation ist aufgrund der Zonulainsuffizienz, schlechter Pupillenerweiterung und Mikrophthalmie schwierig.
Technik: Phakoemulsifikation + IOL-Implantation. Erwägung der Verwendung eines Kapselspannrings (CTR). Ein Pupillendilatationsgerät (Malyugin-Ring usw.) kann erforderlich sein.
Optionen: Gleichzeitige Implantation einer künstlichen Iris (Customflex Artificial Iris usw.) in Betracht ziehen5). Dies kann die Ästhetik verbessern und die Blendungsempfindlichkeit verringern.
Glaukombehandlung
Medikamentöse Therapie: Augentropfen zur Drucksenkung wie Prostaglandinanaloga, Betablocker und Carboanhydrasehemmer verwenden.
Operation: Bei medikamentöser Resistenz Trabekulotomie oder Trabekulektomie (filtrierende Operation) in Betracht ziehen.
Nachsorge: Da Glaukom irreversible Gesichtsfeldausfälle verursacht, sind regelmäßige Augeninnendruckkontrollen und Gesichtsfelduntersuchungen unerlässlich.
Netzhautablösungsbehandlung
Pathologie: Sie kann durch Netzhautrisse innerhalb oder am Rand des Aderhautkoloboms oder durch Risse unabhängig vom Kolobom verursacht werden.
Behandlung: Vitrektomie ist die Grundlage. Die Nützlichkeit einer prophylaktischen Laserphotokoagulation am Rand des Aderhautkoloboms wurde berichtet6).
Prognose: Fälle mit Netzhautablösung haben oft eine schlechte Prognose. Früherkennung und -behandlung sind wichtig.
Regelmäßige Überwachung
Zielkomplikationen: Regelmäßige Nachsorge für die drei Hauptkomplikationen (Glaukom, Katarakt, Netzhautablösung) ist unerlässlich.
Pädiatrisches Management: Regelmäßige Beurteilung von Amblyopie, Strabismus und Augenbewegungsstörungen ist ebenfalls wichtig.
Überwachungshäufigkeit: Je nach Zustand und Alter alle 3 bis 12 Monate augenärztliche Kontrollen fortsetzen.
QKann ein Iriskolobom operativ behoben werden?
A
Es gibt keine Operation, die das Iriskolobom selbst heilt. Ziel der Behandlung ist die symptomatische Therapie der Photophobie und das Management von Komplikationen. Bei begleitendem Katarakt wird eine Phakoemulsifikation mit Intraokularlinsenimplantation durchgeführt, jedoch aufgrund der Schwäche der Zonulafasern mit hohem Risiko. Die künstliche Iris (Customflex Artificial Iris) ist eine wirksame Option zur kosmetischen Verbesserung und Reduktion der Photophobie. Bei Glaukom oder Netzhautablösung werden entsprechende operative Eingriffe durchgeführt.
6. Pathophysiologie und detaillierte Entstehungsmechanismen
Während der embryonalen Augenentwicklung bildet sich nach der Bildung des Augenbechers die Augenbecherspalte (optische Fissur). Um die 6. bis 7. Schwangerschaftswoche beginnt der Verschluss der Spalte am unteren Äquator und schreitet in beide Richtungen zum hinteren Pol und zur Peripherie fort. Wenn dieser Verschluss nicht vollständig erfolgt, entstehen Kolobome in der unteren Uvea (Iris, Ziliarkörper, Aderhaut) und im Sehnerv.
Der Verschluss der Augenbecherspalte erfordert eine koordinierte Steuerung von Basalmembran-Remodellierung, Zellproliferation und Apoptose7). Jede Störung dieses komplexen Prozesses kann zu einem Kolobom führen.
Am Verschluss der Augenbecherspalte sind verschiedene molekulare Signale wie SHH (Sonic Hedgehog), PAX2, BMP und Retinsäure koordiniert beteiligt7). In Tiermodellen wie Zebrafisch und Maus führen Mutationen von pax2, shh und vsx2 zu Kolobomen7). Diese Studien tragen zur Aufklärung des gesamten molekularen Mechanismus des Spaltenverschlusses bei.
Beim Aderhautkolobom fehlen histologisch das Pigmentepithel und die Choriokapillaris, und es liegt eine Dysplasie der undifferenzierten neurosensorischen Netzhaut vor. An den Rändern des Koloboms befindet sich degeneriertes Netzhautgewebe, was ein Risiko für Netzhautforamina darstellt. Diese histologische Eigenschaft ist die pathophysiologische Grundlage für das hohe Risiko einer Netzhautablösung.
Die Sehprognose ist bei isoliertem Iriskolobom oft gut, verschlechtert sich jedoch, je weiter das Aderhautkolobom den hinteren Pol und die Makula einbezieht. Bei beidseitigem Auftreten kann ein Nystagmus auftreten, und es besteht ein Risiko für Amblyopie. Zudem kommt es in vielen Fällen zu einer Netzhautablösung, insbesondere durch Netzhautforamina innerhalb oder entlang des Aderhautkoloboms sowie durch vom Kolobom unabhängige Foramina. Bei Netzhautablösung ist die Prognose oft ungünstig.
Fortschritte in der genetischen Diagnostik: Die gleichzeitige Analyse von Kolobom-assoziierten Genen mittels Next-Generation-Sequencing (NGS)-Panel-Tests wird immer häufiger eingesetzt2). In Zusammenarbeit mit der genetischen Beratung verbessert sich die Genauigkeit der Prognose für die Familie.
Konzept des MAC-Spektrums: Das Konzept eines integrierten Verständnisses von Mikrophthalmie, Anophthalmie und Kolobom als MAC-Spektrum entwickelt sich weiter10). Die genetischen und phänotypischen Überschneidungen der einzelnen Erkrankungen werden zunehmend deutlicher.
Verbreitung der künstlichen Iris: Die Customflex Artificial Iris (HumanOptics) erhielt 2018 die FDA-Zulassung5) und wird als Option zur kosmetischen Verbesserung und Reduzierung von Photophobie bei Irisdefekten eingesetzt. Eine kontinuierliche Sammlung von Anwendungsdaten in Japan wird erwartet.
Gentherapie: Die direkte Gentherapie für Kolobom befindet sich im Stadium der Grundlagenforschung und hat noch keine klinische Anwendung erreicht.
iPS-Zellforschung: Die Grundlagenforschung zur Netzhautregeneration und Irisrekonstruktion mit iPS-Zellen ist im Gange, und eine zukünftige klinische Anwendung wird erwartet.
Morrison D, FitzPatrick D, Hanson I, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39(1):16-22. PMID: 11826019
Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004;41(12):881-891. PMID: 15591273
Pagon RA, Graham JM Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223-227. PMID: 6166737
Mayer CS, Laubichler AE, Khoramnia R, et al. Challenges and complication management in novel artificial iris implantation. J Ophthalmol. 2018;2018:3262068. PMID: 30345111. doi:10.1155/2018/3262068.
Gopal L, Badrinath SS, Kumar KS, et al. Optic disc in fundus coloboma. Ophthalmology. 1996;103(12):2120-2127. PMID: 9003347
Patel A, Sowden JC. Genes and pathways in optic fissure closure. Semin Cell Dev Biol. 2019;91:55-65. doi:10.1016/j.semcdb.2017.10.010. PMID:29198497.
Hornby SJ, Adolph S, Gilbert CE, et al. Visual acuity in children with coloboma: clinical features and a new phenotypic classification system. Ophthalmology. 2000;107(3):511-520. PMID: 10711890
Nakamura KM, Diehl NN, Mohney BG. Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol. 2011;129(1):69-74. PMID: 21220631
Skalicky SE, White AJ, Grigg JR, Martin F, Smith J, Jones M, et al. Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum. JAMA Ophthalmol. 2013;131(12):1517-24. doi:10.1001/jamaophthalmol.2013.5305. PMID:24177921.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.