İris kolobomu (coloboma uveae), embriyonik yarığın kapanmaması sonucu oluşan konjenital bir göz anomalisidir. Defekt, embriyonik yarık boyunca optik disk, koroid, siliyer cisim ve iriste meydana gelir. Optik çanağın alt tarafında oluşan defekte tipik kolobom denir ve diğer bölgelerdeki (makula kolobomu gibi) defektlere atipik kolobom denir.
Tipik iris kolobomu (coloboma iridis), optik çanak yarığının kapanmaması sonucu oluşur. Optik çanak, göz küresinin içeriğini oluşturacak dokuyu yukarıdan aşağıya sarar ve sonunda alt tarafta kapanır. Bu nedenle tipik iris kolobomu alt tarafta (kulak altı tarafına yakın) bulunur ve siliyer cisim kolobomu, koroid kolobomu ve optik sinir kolobomu ile birlikte olabilir. İris kolobomu tek başına da oluşabilir, ancak diğer uvea kolobomları ile birlikteliği sıktır.
İris kolobomunun kendisini ‘iyileştiren’ bir tedavi şu anda mevcut değildir. Tedavi hedefi, fotofobiye yönelik semptomatik tedavi, komplikasyonların yönetimi ve önlenmesi ile görme fonksiyonunun optimize edilmesidir.
Sporadik, otozomal dominant (AD), otozomal resesif (AR) gibi çeşitli
Eşlik eden sendromlar
CHARGE sendromu (CHD7 mutasyonu) gibi çoklu organ anomalilerinin bir belirtisi olarak keşfedilebilir.
Qİris kolobomu (koloboma) kalıtsal mıdır?
A
Kalıtım şekli çeşitlidir; sporadik olgular olabileceği gibi, otozomal dominant (AD) veya resesif (AR) kalıtım gösteren ailelerde birden fazla hasta da bulunabilir. CHARGE sendromu (CHD7 mutasyonu, AD) gibi çoklu organ malformasyon sendromlarının bir parçası olarak da ortaya çıkabilir. Aile öyküsü varlığında veya sendromik olduğundan şüphelenildiğinde genetik danışmanlık önerilir.
Lingam G, et al. Ocular coloboma-a comprehensive review for the clinician. Eye (Lond). 2021. Figure 2. PMCID: PMC8302742. License: CC BY.
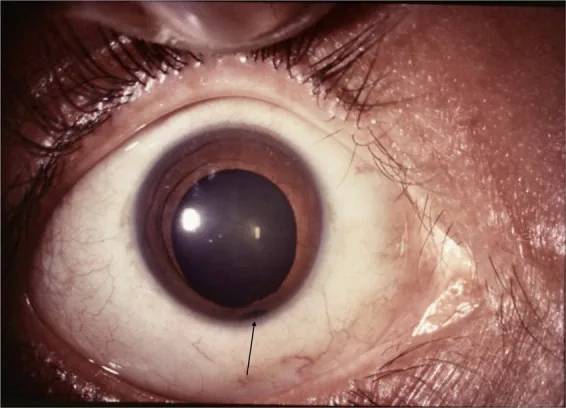
Ön segment fotoğrafında, pupilin alt kısmında anahtar deliği şeklinde bir yarık ile birlikte iris defekti net bir şekilde görülüyor. Bu, iris kolobomunun tipik klinik bulgusunu göstermektedir ve ana belirtiler ile klinik bulguların açıklanması için uygundur.
Görme keskinliği: Tek başına iris kolobomunda görme genellikle iyidir. Defekt makulaya kadar uzanırsa görme keskinliği kötüleşir.
Fotofobi (ışığa hassasiyet): İris defekti nedeniyle ışık miktarının ayarlanamaması fotofobiye yol açar. Günlük yaşamda ciddi rahatsızlığa neden olabilir.
Nistagmus (göz titremesi): Çift taraflı olgularda nistagmus görülebilir. Özellikle görme keskinliği düşük olgularda belirgindir.
Defekt genellikle alt-temporal taraftadır, pupil kenarında en geniştir ve köke doğru daralır (anahtar deliği pupili: keyhole pupil). Sıklıkla mikroftalmi, katarakt ve koroidal kolobom eşlik eder.
İris kolobomu
Yerleşim: Alt (temporal-alt) bölgede sık görülür.
Görünüm: Anahtar deliği şeklinde pupil deformitesi. Pupil kenarında en geniş, köke doğru daralır.
Ana belirtiler: Işık ayar bozukluğuna bağlı fotofobi. Tek başına defektte görme genellikle iyidir.
Siliyer cisim kolobomu
Yerleşim: Genellikle iris defektinin devamı şeklindedir.
Özellik: Tek başına görülmesi nadirdir. Genellikle büyük bir koroid kolobomunun devamıdır. Epitel ve kas defekti mevcuttur.
Komplikasyonlar: Zinn zonüllerinde zayıflama. Lens destek fonksiyonunda azalma.
Koroid kolobomu
Bulgular: Fundusun alt kısmında, sınırları belirgin, çökük, sarı-beyaz bir lezyon olarak gözlenir.
Komplikasyonlar: Görme alanı defekti ve optik atrofi. Görme üzerinde büyük etkisi vardır.
Hornby ve arkadaşlarının fenotipik sınıflamasına göre, kolobomun arka segmente yayılımı görme prognozunu belirleyen ana faktördür ve defekt makulaya ne kadar uzanırsa prognoz o kadar kötüdür8).
Qİris defekti görmeyi nasıl etkiler?
A
Sadece iris defekti varsa çoğu vakada görme iyidir. Ancak koroid kolobomu makulaya yayılırsa görme ciddi şekilde azalır ve prognoz kötüdür. İki taraflı olgularda nistagmus eşlik edebilir ve ambliyopi de bulunabilir. Düzenli göz muayenesi ile görme ve fundus takibi önemlidir.
Embriyonik 6-7. haftalarda fetal yarık (optik fissür/koroidal fissür) kapanması gerçekleşir. Kapanma alt ekvatordan başlar ve arka kutup ile periferik bölgelere doğru çift yönlü ilerler. Bu nedenle arka kutup koroidi ve iris alt kısmında kolobom olarak kalma eğilimi vardır. Kapanma tam olmazsa, alt uvea (iris, siliyer cisim, koroid) ve optik sinirde defektler oluşur.
Çevresel faktörlerle ilgili olarak hayvan deneylerinde teratojenik maddelerin rolü bildirilmiştir, ancak insanlarda kanıtlanmış risk faktörleri sınırlıdır2).
CHARGE sendromu, CHD7 gen mutasyonuna bağlı çoklu organ malformasyon sendromudur. Adı, ana bulgularının baş harflerinden türetilmiştir: Kolobom, Kalp kusurları, Koanal atrezi, Büyüme/gelişme geriliği, Genital anomaliler, Kulak anomalileri. Otozomal dominant kalıtılır ve kolobom sıklıkla tanıya götüren ilk ipucudur. Sistemik yönetim için multidisipliner bakım gereklidir.
Yarık lamba muayenesi: Ön segment muayenesinde alt kadranda iris defekti (anahtar deliği şeklinde pupil) doğrulanır. Kesin tanı için ana bulgudur.
Dilate fundus muayenesi: Koroidal kolobom ve optik sinir kolobomu birlikteliğini doğrulamak için yapılır. Maküler bölgeye yayılım olup olmadığı değerlendirilir.
B-mod ultrasonografi: Mikroftalmiyi doğrulamak ve aksiyel uzunluğu değerlendirmek için yapılır.
OCT (Optik Koherens Tomografi): Maküler bölgeye defekt yayılımı ve maküler hipoplaziyi değerlendirmek için kullanılır.
Göz içi basıncı ölçümü: Glokom birlikteliği için tarama amacıyla yapılır. Düzenli göz içi basıncı ölçümü önemlidir.
Sistemik muayeneler: CHARGE sendromunu dışlamak için ekokardiyografi, işitme testi ve koanal BT yapılır.
Genetik test: CHD7 (CHARGE), PAX2, SOX2 gibi genlerin analizi yapılır.
PAX6 mutasyonu. İris neredeyse tamamen (>2/3) yoktur. Sıklıkla keratopati ve maküler hipoplazi eşlik eder4). Wilms tümörü (WAGR sendromu) riski aniridiye özgüdür ve iris kolobomunda genellikle görülmez.
Travmatik iris diyalizi
Travma öyküsü vardır. Düzensiz iris yırtığı olup oluşum yeri travma bölgesine karşılık gelir.
Pupil bölgesinde membranöz yapı kalır. İris stroması defektli değildir
Qİris defekti ne zaman ve nasıl keşfedilir?
A
Çoğu doğumda veya bebeklik döneminde ön segment muayenesinde anahtar deliği şeklinde pupil olarak keşfedilir. Yarık lamba mikroskobu ile kesin tanı konur ve pupil dilatasyonu ile fundus muayenesi yapılarak koroid ve optik sinir komplikasyonları kontrol edilir. CHARGE sendromundan şüpheleniliyorsa kalp, kulak ve burun için tam sistemik inceleme yapılır. Okul taramaları veya bebek gelişim muayenelerinde tesadüfen de keşfedilebilir.
İris defektinin kendisini tamamen iyileştiren bir tedavi yoktur. Ana semptom olan fotofobiye yönelik semptomatik tedavi, tedavinin başlangıç noktasıdır.
Işık koruyucu gözlükler: İris defektine bağlı ışık ayar bozukluğunu telafi eden ve fotofobiyi azaltan en temel önlem. Koruma derecesi ve renk filtresi semptomlara göre seçilir.
Yapay irisli renkli kontakt lensler (protez tipi KL): Hem kozmetik iyileşme hem de fotofobi azalması sağlar. İris defektini optik olarak tamamlar.
Kırma kusuru düzeltilmesi: Gözlük veya normal kontakt lenslerle kırma kusurunun düzeltilmesi.
Ambliyopi önlenmesi: Çocuklarda uygun kırma kusuru düzeltmesi ve sağlam gözün kapatılması ambliyopi ve şaşılığın önlenmesinde önemlidir.
Takip: Glokomda görme alanı hasarı geri dönüşümsüz olduğundan, düzenli göz içi basıncı izlemi ve görme alanı testi zorunludur.
Retina Dekolmanı Tedavisi
Patofizyoloji: Koroid kolobomu içinde veya kenarında retina yırtığına bağlı veya kolobomla ilişkisiz yırtıklara bağlı olabilir.
Tedavi: Vitrektomi temel tedavidir. Koroid kolobomu kenarına profilaktik lazer fotokoagülasyonun yararlı olduğu bildirilmiştir 6).
Prognoz: Retina dekolmanı gelişen olgularda prognoz genellikle kötüdür. Erken tanı ve tedavi önemlidir.
Düzenli Takip
Hedef komplikasyonlar: Glokom, katarakt ve retina dekolmanı olmak üzere üç ana komplikasyon için düzenli takip zorunludur.
Pediatrik yönetim: Ambliyopi, şaşılık ve oküler motor anormalliklerin düzenli değerlendirilmesi de önemlidir.
Takip sıklığı: Hastalık durumuna ve yaşa bağlı olarak 3-12 ayda bir göz muayenesi sürdürülür.
Qİris kolobomu ameliyatla düzeltilebilir mi?
A
İris kolobomunun kendisini tedavi edecek radikal bir cerrahi yoktur. Tedavinin amacı fotofobiye yönelik semptomatik tedavi ve komplikasyon yönetimidir. Katarakt eşlik ediyorsa fakoemülsifikasyon + göz içi lens implantasyonu yapılır, ancak Zinn zonüllerinin zayıflığı gibi nedenlerle risk yüksektir. Yapay iris (Customflex Artificial Iris) kozmetik iyileşme ve fotofobi azaltmada etkili bir seçenektir. Glokom veya retina dekolmanı eşlik ediyorsa ilgili cerrahi tedaviler uygulanır.
Embriyonik göz gelişiminde, optik kese oluşumundan sonra optik fissür (optic fissure) oluşur. Gebeliğin 6-7. haftaları civarında, optik fissürün kapanması alt ekvatordan başlar ve arka kutup ile perifere doğru çift yönlü ilerler. Bu kapanma tam olarak gerçekleşmezse, alt uvea (iris, siliyer cisim, koroid) ve optik sinirde kolobom oluşur.
Optik fissürün kapanması, bazal membran yeniden şekillenmesi, hücre proliferasyonu ve apoptozun koordineli kontrolünü gerektirir7). Bu karmaşık süreçteki en ufak bir aksama koloboma yol açar.
Optik fissürün kapanmasında SHH (Sonic Hedgehog), PAX2, BMP ve retinoik asit gibi çeşitli moleküler sinyaller koordineli olarak rol oynar7). Zebra balığı ve fare gibi hayvan modellerinde pax2, shh ve vsx2 mutasyonları kolobomu taklit eder7). Bu çalışmalar, optik fissür kapanmasının moleküler mekanizmasının bütün resmini aydınlatmaktadır.
Koroid kolobomunda histolojik olarak pigment epiteli ve koroid kapiller tabakası yoktur ve farklılaşmamış nöral retina displazisi görülür. Kolobom kenarında dejenece retina dokusu bulunur ve retina yırtığı oluşumu riski oluşturur. Bu histolojik özellik, retina dekolmanı yüksek riskinin patofizyolojik temelidir.
İzole iris kolobomunda görme prognozu genellikle iyidir, ancak koroid kolobomu arka kutup ve makulayı ne kadar çok etkilerse prognoz o kadar kötüleşir. Bilateral vakalarda nistagmus görülebilir ve ambliyopi riski de vardır. Ayrıca önemli sayıda vakada retina dekolmanı gelişir; özellikle koroid kolobomu içinde veya kenarı boyunca retina yırtıkları ve kolobomla ilişkisiz retina yırtıklarına bağlıdır. Retina dekolmanı gelişen vakalarda prognoz genellikle kötüdür.
Genetik tanıdaki ilerlemeler: Yeni nesil dizileme (NGS) panel testleri ile kolobomla ilişkili genlerin toplu analizi yaygınlaşmaktadır 2). Genetik danışmanlıkla iş birliği, ailelerin prognoz tahmin doğruluğunu artırmıştır.
MAC spektrumu kavramı: Mikroftalmi (Microphthalmia), Anoftalmi (Anophthalmia) ve Kolobom (Coloboma)‘u birleştiren MAC spektrumu olarak kapsamlı bir şekilde anlama kavramı ilerlemektedir 10). Her bir durumun genetik ve fenotipik örtüşmeleri giderek netleşmektedir.
Yapay irislerin yaygınlaşması: Customflex Artificial Iris (HumanOptics), FDA onayı (2018) almıştır 5) ve iris defektlerinde kozmetik düzeltme ve fotofobi azaltma seçeneği olarak kullanıma sunulmuştur. Japonya’daki kullanım deneyiminin sürekli olarak birikmesi beklenmektedir.
Gen tedavisi: Kolobom için doğrudan gen tedavisi temel araştırma aşamasındadır ve klinik uygulamaya ulaşmamıştır.
iPS hücre araştırmaları: iPS hücreleri kullanılarak retina rejenerasyonu ve iris rekonstrüksiyonu üzerine temel araştırmalar devam etmekte olup, gelecekte klinik uygulamaya geçilmesi beklenmektedir.
Morrison D, FitzPatrick D, Hanson I, et al. National study of microphthalmia, anophthalmia, and coloboma (MAC) in Scotland: investigation of genetic aetiology. J Med Genet. 2002;39(1):16-22. PMID: 11826019
Gregory-Evans CY, Williams MJ, Halford S, Gregory-Evans K. Ocular coloboma: a reassessment in the age of molecular neuroscience. J Med Genet. 2004;41(12):881-891. PMID: 15591273
Pagon RA, Graham JM Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223-227. PMID: 6166737
Mayer CS, Laubichler AE, Khoramnia R, et al. Challenges and complication management in novel artificial iris implantation. J Ophthalmol. 2018;2018:3262068. PMID: 30345111. doi:10.1155/2018/3262068.
Gopal L, Badrinath SS, Kumar KS, et al. Optic disc in fundus coloboma. Ophthalmology. 1996;103(12):2120-2127. PMID: 9003347
Patel A, Sowden JC. Genes and pathways in optic fissure closure. Semin Cell Dev Biol. 2019;91:55-65. doi:10.1016/j.semcdb.2017.10.010. PMID:29198497.
Hornby SJ, Adolph S, Gilbert CE, et al. Visual acuity in children with coloboma: clinical features and a new phenotypic classification system. Ophthalmology. 2000;107(3):511-520. PMID: 10711890
Nakamura KM, Diehl NN, Mohney BG. Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol. 2011;129(1):69-74. PMID: 21220631
Skalicky SE, White AJ, Grigg JR, Martin F, Smith J, Jones M, et al. Microphthalmia, anophthalmia, and coloboma and associated ocular and systemic features: understanding the spectrum. JAMA Ophthalmol. 2013;131(12):1517-24. doi:10.1001/jamaophthalmol.2013.5305. PMID:24177921.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.