Genetik danışmanlık, «doğru genetik bilgi sağlamak» amacıyla verilen bir sağlık hizmetidir. Hastalara ve ailelerine genetik hastalıkların tanısı, kalıtım şekli, tekrarlama riski, उपलब्ध testler ve tedavi seçenekleri hakkında bilgi verir ve bağımsız karar vermeyi destekler. Bu tanım uluslararası düzeyde de kabul görmektedir ve genetik danışmanlığın üç ayağı «bilgi sağlama», «psikolojik destek» ve «karar verme desteği»dir3).
Kalıtsal göz hastalıkları, doğuştan görme bozukluklarının yaklaşık %43’ünü oluşturur1). Çocuklarda kromozom anomalisi görülme sıklığı yaklaşık %0,5-1’dir. Kırmızı-yeşil renk görme bozukluğu, erkeklerin yaklaşık %5’inde ve kadınların yaklaşık %0,2’sinde görülen en yaygın kalıtsal göz hastalıklarından biridir. Retinitis pigmentosa’nın prevalansı yaklaşık 1/4.000 ile 1/5.000 arasındadır ve görme bozukluğunun başlıca nedenlerinden biridir2).
Genetik danışmanlık yalnızca kalıtsal hastalığı olan hastayı değil, hastalığı geliştirebilecek aile bireylerini ve bunu gelecekteki çocuklarına aktarmaktan endişe duyan taşıyıcıları da kapsar. Göz hekimi, tanıyı koyarken genetik uzmanları ve sertifikalı genetik danışmanlarla işbirliği yaparak bilgi sağlar.
QGenetik danışmanlık nereden alınabilir?
A
Üniversite hastanelerinin veya merkez hastanelerin genetik bölümlerinde ya da sertifikalı genetik danışmanların bulunduğu kurumlarda alınabilir. Japon Genetik Danışmanlık Derneği ve Japon İnsan Genetiği Derneği’nin sertifikalı genetik danışman listelerine bakılarak kurumlar bulunabilir. Nanbyo Bilgi Merkezi’nin web sitesi de danışmanlık yerleri hakkında bilgi sağlar.
2. Kalıtım biçimleri ve başlıca kalıtsal göz hastalıkları
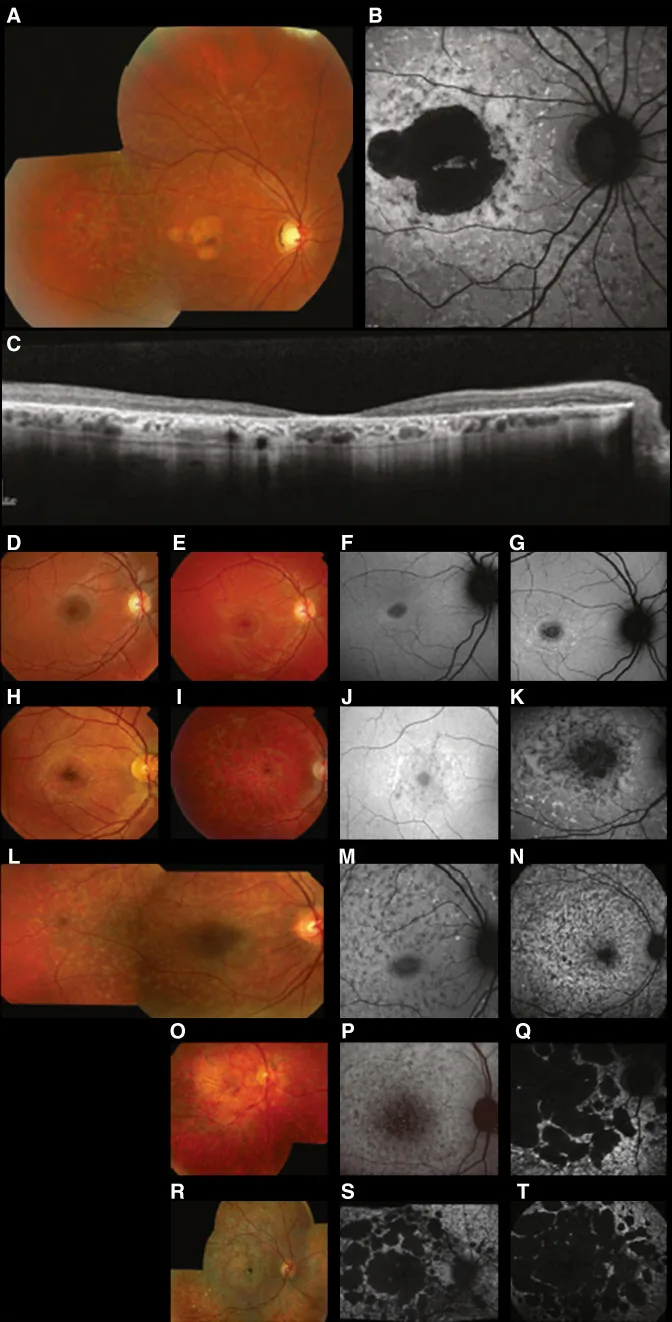
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Stargardt hastalığında (STGD1) tipik çok modlu görüntüleme: renkli fundus fotoğrafı, retina pigment epiteli düzeyinde sarı-beyaz lekeleri ve makula atrofisini gösterir (A); fundus otofloresansı, makulada düşük floresanlı bir alanı ve çevresindeki anormal floresansı gösterir (B); SD-OCT ise dış retina katmanları ve RPE’nin belirgin kaybını ve lekelerle uyumlu hiperreflektif odakları gösterir (C). Bu, “2. Kalıtım biçimleri ve başlıca kalıtsal göz hastalıkları” bölümünde ele alınan Stargardt hastalığına (ABCA4 gen mutasyonlarına bağlı otozomal resesif retinal distrofi) karşılık gelir.
Kalıtım paterni, genetik danışmanlıkta temel bir bilgidir ve dört ana paterne ayrılır.
Otozomal dominant kalıtım
Ortaya çıkma koşulu: Hastalık, tek bir allelde mutasyon olduğunda ortaya çıkar (heterozigot durum).
Soy ağacı özellikleri: Etkilenen bireyler ardışık nesillerde görülür.
Yineleme riski: Bir çocuğa geçirme olasılığı %50’dir.
Not: Penetrans %100 değilse bir nesil atlanmış gibi görünebilir.
Otozomal resesif kalıtım
Ortaya çıkma koşulu: Her iki allel de mutasyona uğradığında hastalık ortaya çıkar (homozigot veya bileşik heterozigot).
Soy ağacı özellikleri: Etkilenen bireyler kardeşler arasında görülür. Ebeveynler genellikle taşıyıcıdır (heterozigot).
Yineleme riski: İki taşıyıcıdan doğan bir শিশekte hastalık olasılığı %25’tir.
Son eğilim: Kuzen evliliklerinin azalması nedeniyle bileşik heterozigotların oranı artmıştır.
X’e bağlı kalıtım
Ortaya çıkma koşulu: Hastaların çoğu erkektir (hemizigot).
Kadınlarda: İki X kromozomu olduğu için, bunlardan birinde mutasyon olursa taşıyıcı olur.
Aile ağacı özellikleri: Etkilenenler daha çok erkeklerdir ve aktarım anneden oğula olur.
Yeniden görülme riski: Taşıyıcı bir annenin oğlunun etkilenme olasılığı %50’dir.
Anne tarafından kalıtım (mitokondriyal kalıtım)
Özellik: Spermdeki mitokondriyal DNA (mtDNA), döllenme sırasında neredeyse tamamen parçalanır. Bu nedenle yalnızca anneden çocuğa aktarılır.
QEbeveynlerden birinde kalıtsal bir göz hastalığı varsa, bunun çocuğa geçme olasılığı nedir?
A
Kalıtım modeli farklıdır. Otozomal dominant kalıtımda, etkilenmiş bir ebeveynden çocuğa geçme olasılığı %50’dir. Otozomal resesif kalıtımda, her iki ebeveyn de taşıyıcıysa çocuğun hastalığı geliştirme olasılığı %25’tir. X’e bağlı kalıtımda, taşıyıcı bir annenin oğlunun etkilenme olasılığı %50’dir ve anne yoluyla kalıtımda (mitokondriyal), anne hasta ya da taşıyıcıysa tüm çocuklara geçebilir. Bireysel durumlar için genetik uzmanına veya sertifikalı genetik danışmana başvurulması önerilir.
3. Kalıtsal göz hastalıklarının nedenleri ve risk faktörleri
Kalıtsal göz hastalıklarının ortaya çıkış mekanizmaları, genetik varyantın protein işlevi üzerindeki etkisinin türüne göre sınıflandırılır.
Haployetersizlik (haploinsufficiency): Yalnızca bir alelin işlevini kaybetmesiyle fenotip ortaya çıkar. Klasik örnek doğuştan anirididir (PAX6 mutasyonu). Tek bir normal alel, normal doku oluşumu için gereken gen ürününün miktarını karşılayamaz.
Dominant negatif etki (dominant negative effect): Mutant protein, normal proteinin işlevini hem rekabetçi hem de yapısal olarak engeller. Marfan sendromunda (FBN1 mutasyonu) mutant fibrillin-1 molekülleri hücre dışı matriksin oluşumunu bozar.
Mitokondriyal mutasyonlar: Leber kalıtsal optik nöropatisinde (LHON), 11778, 3460 ve 14484 numaralı üç nokta mutasyonu tüm mutasyonların yaklaşık %90’ını oluşturur. Enerji üretimindeki bozukluk retina gangliyon hücrelerine zarar verir.
De novo mutasyonlar: Ebeveynlerde bulunmayan ve yumurta ya da sperm oluşumu sırasında yeni ortaya çıkan mutasyonlardır. Bunun tipik bir örneği CRX mutasyonlarına bağlı Leber konjenital amaurozudur. Soy ağacında başka etkilenen kimse olmadığında tekil olgu olarak da görülebilir.
Bileşik heterozigot: Otozomal resesif hastalıklarda, iki alelde iki farklı mutasyonun bulunduğu formdur. Son yıllarda kuzen evliliklerinin azalmasıyla birlikte, homozigotlara kıyasla bileşik heterozigotların oranı artmıştır.
Tek ebeveynli disomi: Aynı kromozomun iki kopyasının tek bir ebeveynden kalıtıldığı, diğer ebeveyne ait kromozomun eksik olduğu bir durumdur. Görünüşte sporadik bir olgu olarak ortaya çıkabilir10).
Retinitis pigmentosa için 100’den fazla neden gen tanımlanmıştır ve aynı fenotip bile çok sayıda farklı gen tarafından oluşturulabilir2). Bu genetik çeşitlilik tanıyı zorlaştırır.
4. Genetik danışmanlık ve genetik testin pratik yönleri
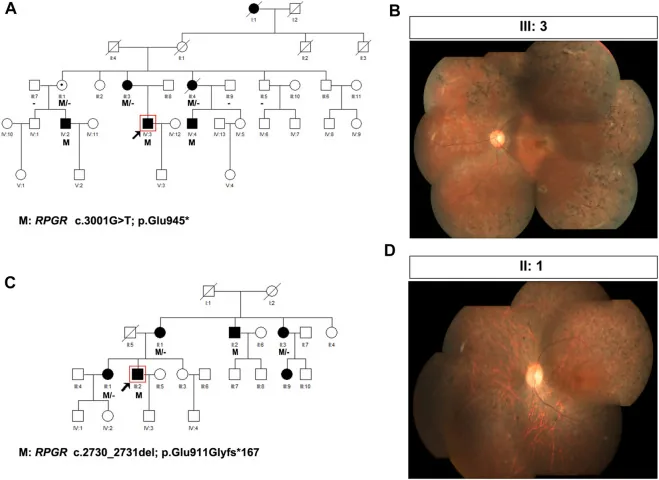
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
X’e bağlı retinitis pigmentosa (RPGR varyantları: c.3001G>T ve c.2730_2731del) olan iki aileye ait soy ağaçları ve fundus fotoğrafları. Soy ağaçlarında kalıtım modelini göstermek için standart semboller (etkilenen erkekler için dolu kareler, taşıyıcı kadınlar için noktalı daireler) kullanılmıştır ve karşılık gelen fundus fotoğraflarında ileri derecede kemiksi pigment birikimi ve retina atrofisi görülmektedir. Bu, “4. Genetik danışmanlığın uygulaması ve genetik testler” bölümünde ele alınan soy ağacı (pedigree) oluşturma ve kalıtım modelinin belirlenmesine karşılık gelir.
Genetik danışmanlığı uygun şekilde uygulamak için aşağıdaki hazırlıklar gerekir.
Soy ağacı (pedigree) oluşturma: En az üç kuşağı kapsayan aile öyküsü alın ve soy ağacı olarak kaydedin. Hastaların dikey ve yatay dağılımı, kalıtım modelinin tahmin edilmesini sağlar.
Kalıtım modelini tahmin etme: Soy ağacı ve fenotipe dayanarak bunun otozomal dominant, otozomal resesif, X’e bağlı veya maternal olup olmadığını tahmin edin.
Genetik testin açıklanması ve yazılı onam alınması: Hastanın genetik bilgileri incelenmeden önce, testin anlamı ve sorunları yeterince açıklanmalı ve yazılı onam alınmalıdır.
DNA testi sonuçlarının anonimleştirilmesi ve bilgi yönetimi: Bağlantı kurulabilir anonimleştirme ile gizliliği koruyun.
Tesadüfi bulgulara nasıl yaklaşılacağının önceden belirlenmesi: Testten önce, yaşamı tehdit edebilecek ciddi tesadüfi bulgular saptanırsa bunların nasıl bildirileceğini hastayla netleştirin.
Yüksek doğruluk. Bilinen mutasyon bölgelerini doğrulamak için uygundur
Panel testi (hedefe yönelik dizileme)
Belirli hastalıklarla ilişkili gen grubu
Retina hastalıklarıyla ilişkili genlerin toplu taranması. Tanı oranı yüksektir5)
Ekzom analizi (NGS)
Tüm ekzon bölgeleri
Bilinmeyen varyantları saptar. Hastalığa neden olan çok sayıda gen olduğunda etkilidir
Tüm genom analizi (WGS)
Tüm genom
Mevcut hedefli NGS testlerine göre daha yüksek tanı oranına sahip olabilir13)
Kalıtsal retina distrofisinde, RPE65 gen mutasyonuna bağlı bir hastalıktan şüphelenildiğinde ve gen tedavisine uygunluğun belirlenmesine yardımcı olmak gibi amaçlarla, koşulları karşılayan bazı testler sigorta kapsamındaki sağlık hizmeti olarak yapılabilir4).
Varyantların yorumlanması ve başlıca veri tabanları
Varyantların yorumlanmasında dikkatli değerlendirme gerekir. Makalelerde “varyant” olarak yayımlanan açıklamaların yaklaşık %30’unun aslında polimorfizm (normal varyant) olduğu bilinmektedir. Genel olarak, sağlıklı her 100 kişiden 1 veya daha fazlasında görülen baz dizisi değişiklikleri polimorfizm olarak değerlendirilmelidir.
Aşağıdaki başlıca veri tabanları kullanılır.
OMIM (Online Mendelian Inheritance in Man): kalıtsal hastalıklar ve genler için kapsamlı veri tabanı
GeneReviews: her hastalık için genetik danışmanlık bilgileri sunar
RetNet (Retinal Information Network): retina hastalıklarına özel gen veri tabanı
QGenetik testin ücreti sigorta kapsamında mı?
A
Bazı kalıtsal göz hastalıklarında, tedavi endikasyonu belirlendiğinde ve belirlenmiş kurum kriterleri karşılandığında genetik test sigorta kapsamındaki sağlık hizmeti olarak alınabilir. Ancak kapsam, testin türüne ve ilgili hastalığa göre değişir; bu nedenle başvurulan kurumda teyit edilmesi gerekir. Sigorta kapsamına girmeyen testler cepten ödemeli olabilir. Belirlenmiş güç tedavi edilen hastalıklarda (örneğin retinitis pigmentosa) bu tür hastalıklar için tıbbi gider destek sistemi aracılığıyla mali destek de sağlanabilir4).
5. Genetik danışmanlık sistemi ve tedaviye ilişkin beklentiler
Genetik danışmanlık yürütülürken, genetik uzmanı ve sertifikalı genetik danışman ile iş birliği ideal kabul edilir. Üniversite hastanelerinde etik kurul incelemesi gerekebilir. Genetik bilgi yalnızca hastayı değil, aile üyelerini de etkileyebileceği için, bu bilgilerin yönetiminde özel dikkat gerekir. Genetik bilgiye dayalı haksız ayrımcılığın önlenmesine ilişkin toplumsal tartışmalar da ilerlemektedir8).
Belirlenmiş nadir hastalıklar ve tıbbi gider desteği
Nadir hastalığı olan hastalarda tıbbi giderlerin cepten ödenen kısmı azaltılır. Ayaktan tedavi, yatış ve ilaçlar için birlikte hesaplanan aylık bir üst sınır belirlenmiştir ve gelire göre kategoriler uygulanır.
RPE65 gen mutasyonlarına bağlı kalıtsal retina distrofisi için gen tedavisi ilacı voretigene neparvovec, AAV2 vektörü kullanılan subretinal uygulamalı bir preparattır. Etkinlik ve güvenlilik randomize kontrollü çalışmalarda doğrulanmıştır6). Tedavi endikasyonunun belirlenmesi için genetik test ile hastalık tipinin doğrulanması gerekir.
Antisens oligonükleotid (ASO) tedavisi açısından, CEP290 mutasyonlarının neden olduğu Leber konjenital amaurozu tip 10 (LCA10) için bir preparatın intravitreal uygulaması klinik çalışmalarda araştırılmaktadır9).
iPS hücreleri kullanılarak yapılan otolog retinal pigment epiteli hücre nakli, yaşa bağlı makula dejenerasyonu için rejeneratif tıp olarak araştırılmaktadır7). Kalıtsal retina hastalıklarından ayrı bir alan olsa da, retina hücre tedavisinin somut bir örneği olarak dikkat çekmektedir.
QGen tedavisi şimdi uygulanabiliyor mu?
A
RPE65 mutasyonuna bağlı kalıtsal retinal distrofi için voretigene neparvovec’in etkililiği ve güvenliği RKT’lerde6) gösterilmiştir. Tedavi uygunluğunu doğrulamak için, hastalık tipinin genetik testle belirlenmesi gerekir. CEP290 mutasyonlu LCA10’da ASO preparatlarının intravitreal enjeksiyonu klinik çalışmalarda araştırılmaktadır9).
Otozomal dominant, otozomal resesif ve X’e bağlı kalıtım Mendel yasalarına uyar. Ancak aşağıdaki faktörler basit tahmini zorlaştırabilir.
Penetrans: Mutasyon taşısa bile herkes hastalanmaz. Penetrans düşük olduğunda özellik nesil atlayabilir ve soyağacından kalıtım paternini çıkarmak zorlaşır.
Ekspresivite: Aynı mutasyona sahip aile bireyleri arasında bile belirtilerin şiddeti farklı olabilir.
Kazanım-fonksiyon mutasyonu (gain-of-function): Mutant proteinin yeni ve zararlı bir işlev kazandığı mekanizma. Bu, olağan dominant-negatif etkiden farklıdır.
Mitokondriler sitoplazmada bulunur ve çocuğa yalnızca annenin yumurtasından gelen mtDNA aktarılır. Her hücrede binlerce mtDNA kopyası vardır ve mutasyona uğramış mtDNA ile normal mtDNA’nın birlikte bulunduğu bir durum (heteroplazmi) ortaya çıkabilir. Heteroplazmi oranı ne kadar yüksekse, belirtiler genellikle o kadar ağır olur. Leber herediter optik nöropatisinde (LHON), 11778 (en sık), 3460 ve 14484 olmak üzere üç varyant tüm varyantların yaklaşık %90’ını oluşturur.
Tek ebeveynli disomi, bir kromozom çiftinin her iki kromozomunun da aynı ebeveynden alınması ve diğer ebeveyne ait kromozomun bulunmaması durumudur. Otozomal resesif bir hastalığın taşıyıcısından doğan bir çocuk, diğer ebeveyn taşıyıcı olmasa bile hastalığa yakalanabilir ve bu durum görünüşte sporadik bir olgu gibi olabilir10). De novo varyantlar ve bileşik heterozigotlarla birlikte, aile öyküsü olmasa bile kalıtsal hastalık düşünülmelidir.
7. En son araştırmalar ve geleceğe yönelik beklentiler
CRISPR/Cas9 kullanan gen düzenleme tedavi adayı EDIT-101’in, LCA10 (CEP290 varyantları) için preklinik geliştirme aşamasında olduğu bildirilmiştir11).
Preimplantasyon genetik test (PGT-M), otozomal dominant ve resesif kalıtsal hastalıklar için uygulanabilir ve etik bir çerçevede değerlendirilebilir12).
Tüm genom dizilemenin (WGS), kalıtsal retina hastalıklarında mevcut standart genetik testlere kıyasla moleküler tanı oranını artırabileceği gösterilmiştir13).
Gen varyantlarının patojenitesini tahmin etmeye yönelik yapay zekâ (AI) tabanlı araçlar geliştirilmektedir ve varyant yorumlama doğruluğunun artması beklenmektedir.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.