Tư vấn di truyền là một dịch vụ y tế nhằm mục đích «cung cấp thông tin di truyền chính xác». Dịch vụ này cung cấp cho bệnh nhân và gia đình thông tin về chẩn đoán, kiểu di truyền, nguy cơ tái phát, các xét nghiệm hiện có và phương pháp điều trị của bệnh di truyền, đồng thời hỗ trợ họ tự ra quyết định. Định nghĩa này cũng được quốc tế công nhận, và ba trụ cột của tư vấn di truyền là «cung cấp thông tin», «hỗ trợ tâm lý» và «hỗ trợ ra quyết định»3).
Các bệnh mắt di truyền chiếm khoảng 43% các trường hợp suy giảm thị lực bẩm sinh1). Tần suất bất thường nhiễm sắc thể ở con cái khoảng 0,5–1%. Rối loạn nhìn màu đỏ-xanh là một trong những bệnh mắt di truyền thường gặp nhất, xuất hiện ở khoảng 5% nam giới và 0,2% nữ giới. Tỷ lệ hiện mắc của thoái hóa sắc tố võng mạc khoảng 1/4.000 đến 1/5.000, và đây là một trong những nguyên nhân chính gây suy giảm thị lực2).
Đối tượng của tư vấn di truyền không chỉ là chính bệnh nhân mắc bệnh di truyền, mà còn bao gồm các thành viên gia đình có thể mắc bệnh và những người mang gen lo lắng về việc truyền bệnh cho con trong tương lai. Bác sĩ nhãn khoa, trong khi đảm nhiệm chẩn đoán, cũng phối hợp với bác sĩ chuyên khoa di truyền và tư vấn viên di truyền được chứng nhận để cung cấp thông tin.
QTư vấn di truyền có thể được thực hiện ở đâu?
A
Có thể được thực hiện tại khoa di truyền của bệnh viện đại học hoặc bệnh viện đầu mối, hoặc tại cơ sở có tư vấn viên di truyền được chứng nhận. Có thể tìm cơ sở bằng cách tham khảo danh sách tư vấn viên di truyền được chứng nhận của Hội Tư vấn Di truyền Nhật Bản và Hội Di truyền Người Nhật Bản. Trang web của Trung tâm Thông tin Bệnh Nan y cũng cung cấp hướng dẫn nơi tư vấn.
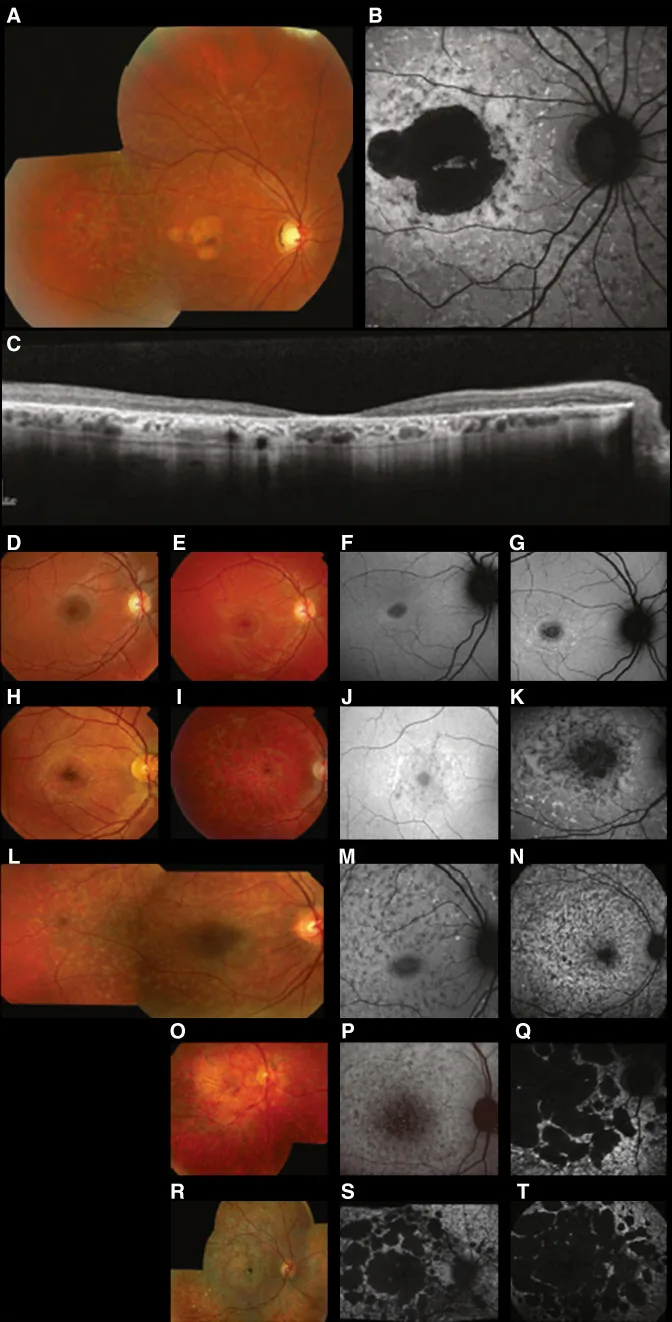
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Hình ảnh đa phương thức điển hình của bệnh Stargardt (STGD1): ảnh màu đáy mắt cho thấy các đốm trắng vàng ở mức biểu mô sắc tố võng mạc và teo hoàng điểm (A); chụp tự phát quang đáy mắt cho thấy vùng giảm phát quang ở hoàng điểm với phát quang bất thường xung quanh (B); và SD-OCT cho thấy mất rõ các lớp võng mạc ngoài và RPE, cùng các ổ tăng phản xạ tương ứng với các đốm (C). Đây là bệnh Stargardt (loạn dưỡng võng mạcdi truyền lặn trên nhiễm sắc thể thường do đột biến gen ABCA4) được trình bày trong mục “2. Kiểu di truyền và các bệnh mắt di truyền chính”.
Kiểu di truyền là thông tin cốt lõi trong tư vấn di truyền và được phân thành bốn kiểu chính.
Di truyền trội trên nhiễm sắc thể thường
Điều kiện khởi phát: Bệnh xuất hiện khi có đột biến ở một alen (trạng thái dị hợp).
Đặc điểm phả hệ: Các thành viên bị bệnh xuất hiện ở các thế hệ liên tiếp.
Nguy cơ tái phát: Xác suất truyền sang con là 50%.
Lưu ý: Nếu độ thấm không phải 100%, có thể có hiện tượng bỏ qua một thế hệ.
Di truyền lặn trên nhiễm sắc thể thường
Điều kiện khởi phát: Bệnh xuất hiện khi cả hai alen đều bị đột biến (đồng hợp hoặc dị hợp tử kép).
Đặc điểm phả hệ: Các thành viên bị bệnh xuất hiện giữa các anh chị em ruột. Cha mẹ thường là người mang gen bệnh (dị hợp).
Nguy cơ tái phát: Xác suất mắc bệnh ở con của hai người mang gen bệnh là 25%.
Xu hướng gần đây: Do hôn nhân giữa anh em họ ngày càng ít, tỷ lệ dị hợp tử kép đã tăng lên.
Di truyền liên kết X
Điều kiện khởi phát: Phần lớn bệnh nhân là nam (bán hợp tử).
Ở nữ giới: Vì có hai nhiễm sắc thể X, nếu một nhiễm sắc thể bị đột biến thì sẽ trở thành người mang gen bệnh.
Đặc điểm phả hệ: Người mắc bệnh chủ yếu là nam giới, và bệnh được truyền từ mẹ sang con trai.
Nguy cơ tái phát: Xác suất con trai của người mẹ mang gen bệnh mắc bệnh là 50%.
Di truyền theo dòng mẹ (di truyền ty thể)
Đặc điểm: DNA ty thể (mtDNA) của tinh trùng gần như bị phân hủy hoàn toàn khi thụ tinh. Vì vậy, bệnh chỉ được truyền từ mẹ sang con.
QNếu một trong hai cha mẹ mắc bệnh mắt di truyền, khả năng truyền sang con là bao nhiêu?
A
Kiểu di truyền khác nhau. Với di truyền trội trên nhiễm sắc thể thường, khả năng truyền từ cha hoặc mẹ bị bệnh sang con là 50%. Với di truyền lặn trên nhiễm sắc thể thường, nếu cả hai cha mẹ đều là người mang gen, khả năng con mắc bệnh là 25%. Với di truyền liên kết nhiễm sắc thể X, con trai của người mẹ mang gen có 50% khả năng mắc bệnh, và với di truyền theo dòng mẹ (ty thể), nếu người mẹ mắc bệnh hoặc là người mang gen, bệnh có thể truyền cho tất cả con. Với từng trường hợp cụ thể, nên trao đổi với bác sĩ chuyên khoa di truyền hoặc chuyên viên tư vấn di truyền được chứng nhận.
3. Nguyên nhân và yếu tố nguy cơ của bệnh mắt di truyền
Cơ chế khởi phát của bệnh mắt di truyền được phân loại theo kiểu tác động mà đột biến gen gây ra lên chức năng protein.
Thiếu hụt đơn alen (haploinsufficiency): Kiểu hình xuất hiện chỉ với mất chức năng của một alen. Ví dụ điển hình là vô mống mắt bẩm sinh (đột biến PAX6). Chỉ một alen bình thường không thể cung cấp đủ sản phẩm gen cần cho sự hình thành mô bình thường.
Hiệu ứng trội âm (dominant negative effect): Protein đột biến ức chế chức năng của protein bình thường theo kiểu cạnh tranh và về cấu trúc. Trong hội chứng Marfan (đột biến FBN1), các phân tử fibrillin-1 đột biến làm rối loạn sự hình thành chất nền ngoại bào.
Đột biến ty thể: Trong bệnh thần kinh thị giác di truyền Leber (LHON), ba đột biến điểm 11778, 3460 và 14484 chiếm khoảng 90% tổng số đột biến. Rối loạn tạo năng lượng làm tổn thương các tế bào hạch võng mạc.
Đột biến de novo: Đột biến không có ở cha mẹ và phát sinh mới trong quá trình hình thành trứng hoặc tinh trùng. Một ví dụ điển hình là mù bẩm sinh Leber do đột biến CRX. Chúng cũng có thể xuất hiện dưới dạng ca đơn lẻ khi không có ai khác trong phả hệ bị ảnh hưởng.
Dị hợp tử kép: Trong bệnh lặn trên nhiễm sắc thể thường, đây là dạng mà hai alen mang hai đột biến khác nhau. Cùng với sự giảm dần của hôn nhân cận huyết gần đây, tỷ lệ dị hợp tử kép đã tăng lên so với đồng hợp tử.
Đồng nhiễm sắc thể từ một bên cha mẹ: Hiện tượng trong đó cả hai bản sao của cùng một nhiễm sắc thể đều được di truyền từ một bên cha hoặc mẹ, trong khi nhiễm sắc thể từ bên còn lại không có. Nó có thể xuất hiện như một ca tưởng như lẻ tẻ10).
Đã xác định được hơn 100 gen gây bệnh võng mạc sắc tố, và ngay cả cùng một kiểu hình cũng có thể do nhiều gen khác nhau gây ra2). Sự đa dạng di truyền này khiến việc chẩn đoán trở nên khó khăn.
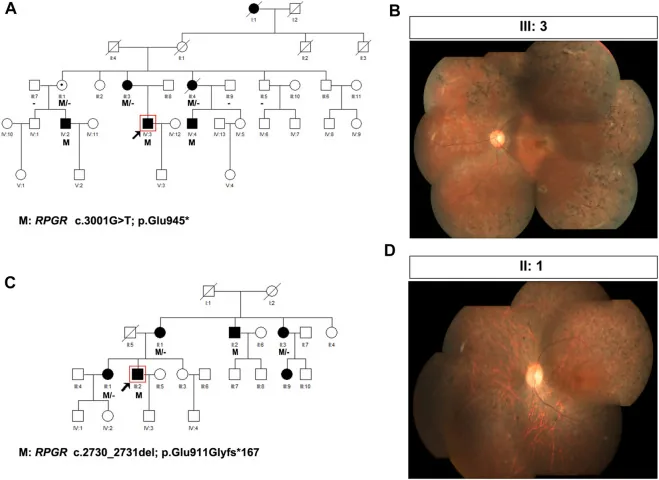
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Phả hệ và ảnh đáy mắt của hai gia đình mắc bệnh thoái hóa sắc tố võng mạc liên kết X (biến thể RPGR: c.3001G>T và c.2730_2731del). Phả hệ sử dụng các ký hiệu chuẩn (hình vuông tô kín cho nam giới bị bệnh, hình tròn nét chấm cho nữ mang gen) để thể hiện kiểu di truyền, và các ảnh đáy mắt tương ứng cho thấy sắc tố dạng gai xương tiến triển và teo võng mạc. Điều này tương ứng với việc lập phả hệ và xác định kiểu di truyền được đề cập trong mục “4. Thực hành tư vấn di truyền và xét nghiệm gen”.
Để thực hiện tư vấn di truyền phù hợp, cần chuẩn bị những điều sau.
Lập phả hệ (pedigree): Hỏi tiền sử gia đình ít nhất ba thế hệ và ghi lại dưới dạng phả hệ. Phân bố người mắc theo chiều dọc và chiều ngang có thể giúp ước đoán kiểu di truyền.
Ước đoán kiểu di truyền: Dựa vào phả hệ và kiểu hình, suy ra là trội trên nhiễm sắc thể thường, lặn trên nhiễm sắc thể thường, liên kết X hay di truyền từ mẹ.
Giải thích xét nghiệm gen và lấy đồng ý bằng văn bản: Trước khi xem xét thông tin di truyền của bệnh nhân, cần giải thích đầy đủ ý nghĩa và các vấn đề của xét nghiệm, đồng thời lấy đồng ý bằng văn bản.
Ẩn danh kết quả xét nghiệm DNA và quản lý thông tin: Bảo vệ quyền riêng tư bằng cách ẩn danh có thể liên kết.
Quyết định trước cách xử lý phát hiện tình cờ: Trao đổi trước với bệnh nhân về cách thông báo nếu phát hiện những tình cờ nghiêm trọng có thể đe dọa tính mạng.
Độ chính xác cao. Phù hợp để xác nhận các vị trí đột biến đã biết
Xét nghiệm panel (giải trình tự mục tiêu)
Nhóm gen liên quan đến các bệnh cụ thể
Sàng lọc đồng thời các gen liên quan đến bệnh võng mạc. Tỷ lệ chẩn đoán cao5)
Phân tích exome (NGS)
Toàn bộ vùng exon
Phát hiện các biến thể chưa biết. Hữu ích khi có nhiều gen gây bệnh
Phân tích toàn bộ hệ gen (WGS)
Toàn bộ hệ gen
Có thể có tỷ lệ chẩn đoán cao hơn các xét nghiệm NGS nhắm mục tiêu hiện có13)
Trong loạn dưỡng võng mạc di truyền, khi nghi ngờ bệnh do đột biến gen RPE65, và trong các trường hợp như nhằm hỗ trợ quyết định chỉ định liệu pháp gen, một số xét nghiệm đáp ứng điều kiện có thể được thực hiện như dịch vụ y tế được bảo hiểm chi trả4).
Cần thận trọng khi diễn giải biến thể. Người ta biết rằng khoảng 30% các mô tả được công bố là “biến thể” trong các bài báo thực ra là đa hình (biến thể bình thường). Nói chung, những thay đổi trình tự base xuất hiện ở 1 người trở lên trong mỗi 100 người khỏe mạnh nên được xem là đa hình.
Các cơ sở dữ liệu chính được sử dụng gồm sau đây.
OMIM (Online Mendelian Inheritance in Man): cơ sở dữ liệu toàn diện về bệnh di truyền và gen
GeneReviews: cung cấp thông tin tư vấn di truyền cho từng bệnh
RetNet (Retinal Information Network): cơ sở dữ liệu gen chuyên về các bệnh võng mạc
QChi phí xét nghiệm gen có được bảo hiểm chi trả không?
A
Ở một số bệnh mắt di truyền, xét nghiệm di truyền có thể được thực hiện như dịch vụ y tế được bảo hiểm chi trả khi xác định chỉ định điều trị và đáp ứng các tiêu chí của cơ sở được chỉ định. Tuy nhiên, việc có được chi trả hay không phụ thuộc vào loại xét nghiệm và bệnh được xét, vì vậy cần xác nhận tại cơ sở khám chữa bệnh. Các xét nghiệm không được bảo hiểm chi trả có thể phải tự thanh toán. Đối với các bệnh khó chữa được chỉ định (như viêm sắc tố võng mạc), cũng có thể có hỗ trợ tài chính thông qua chế độ trợ cấp chi phí y tế cho bệnh khó chữa được chỉ định4).
5. Hệ thống tư vấn di truyền và triển vọng điều trị
Khi thực hiện tư vấn di truyền, lý tưởng nhất là phối hợp với bác sĩ chuyên khoa di truyền và chuyên viên tư vấn di truyền được chứng nhận. Ở bệnh viện đại học, có thể cần được hội đồng đạo đức thẩm định. Vì thông tin di truyền có thể ảnh hưởng không chỉ đến người bệnh mà còn đến các thành viên gia đình, cần đặc biệt cẩn trọng trong việc xử lý thông tin. Cuộc thảo luận xã hội về việc ngăn chặn sự phân biệt đối xử không công bằng dựa trên thông tin di truyền cũng đang được thúc đẩy8).
Các bệnh hiếm được chỉ định và hỗ trợ chi phí y tế
Ở bệnh nhân mắc bệnh hiếm, phần chi phí y tế phải tự trả được giảm. Có mức trần hằng tháng cho tổng chi phí khám ngoại trú, nhập viện và thuốc kê đơn, và áp dụng các mức theo thu nhập.
Thuốc liệu pháp gen voretigene neparvovec cho loạn dưỡng võng mạc di truyền do đột biến gen RPE65 là một chế phẩm dùng tiêm dưới võng mạc với vector AAV2. Hiệu quả và độ an toàn đã được xác nhận trong các thử nghiệm ngẫu nhiên có đối chứng6). Việc quyết định chỉ định điều trị đòi hỏi phải xác định loại bệnh bằng xét nghiệm gen.
Đối với liệu pháp oligonucleotide kháng nghĩa (ASO), việc tiêm nội nhãn một chế phẩm dành cho chứng amaurosis bẩm sinh Leber тип 10 (LCA10) do đột biến CEP290 gây ra đang được nghiên cứu trong các thử nghiệm lâm sàng9).
Ghép tự thân tế bào biểu mô sắc tố võng mạc bằng tế bào iPS đang được nghiên cứu như một liệu pháp tái tạo cho thoái hóa hoàng điểm do tuổi tác7). Mặc dù đây là một lĩnh vực khác với bệnh võng mạc di truyền, nó được chú ý như một ví dụ thực tế về liệu pháp tế bào võng mạc.
QHiện nay liệu pháp gen đã có sẵn chưa?
A
Ở loạn dưỡng võng mạc di truyền do đột biến RPE65, hiệu quả và độ an toàn của voretigene neparvovec đã được chứng minh trong các thử nghiệm ngẫu nhiên có đối chứng6). Để xác định có phù hợp điều trị hay không, cần xác định loại bệnh bằng xét nghiệm gen. Ở LCA10 có đột biến CEP290, tiêm nội nhãn các chế phẩm ASO đang được nghiên cứu trong các thử nghiệm lâm sàng9).
Di truyền trội trên nhiễm sắc thể thường, lặn trên nhiễm sắc thể thường và liên kết X tuân theo các quy luật của Mendel. Tuy nhiên, các yếu tố sau có thể khiến việc dự đoán đơn giản trở nên khó khăn.
Độ thấm (penetrance): dù có mang đột biến, không phải ai cũng mắc bệnh. Khi độ thấm thấp, bệnh có thể bỏ qua thế hệ, khiến khó suy ra kiểu di truyền từ phả hệ.
Mức độ biểu hiện (expressivity): ngay cả trong gia đình có cùng gen đột biến, mức độ nặng của triệu chứng cũng có thể khác nhau.
Đột biến tăng chức năng (gain-of-function): cơ chế trong đó protein đột biến có được chức năng có hại mới. Điều này khác với hiệu ứng trội âm tính thường gặp.
Ty thể nằm trong bào tương, và chỉ mtDNA có nguồn gốc từ trứng của người mẹ được truyền sang con. Mỗi tế bào có hàng nghìn bản sao mtDNA, và có thể xảy ra tình trạng mtDNA đột biến và mtDNA bình thường cùng tồn tại (heteroplasmy). Tỷ lệ heteroplasmy càng cao thì triệu chứng thường càng nặng. Ở bệnh mù thần kinh thị giác di truyền Leber (LHON), ba biến thể—11778 (thường gặp nhất), 3460 và 14484—chiếm khoảng 90% tổng số biến thể.
Đơn thân lưỡng bội là tình trạng cả hai nhiễm sắc thể của một cặp đều đến từ cùng một người cha hoặc mẹ, không có nhiễm sắc thể từ người còn lại. Trẻ sinh ra từ một người mang bệnh lặn trên nhiễm sắc thể thường có thể mắc bệnh ngay cả khi người còn lại không phải là người mang gen, nên trông như một trường hợp lẻ tẻ10). Cùng với đột biến de novo và dị hợp tử kép, cần nghĩ đến bệnh di truyền ngay cả khi không có tiền sử gia đình.
Ở LCA10 có biến thể CEP290, việc sửa chữa splicing bằng liệu pháp ASO đang được nghiên cứu trong các thử nghiệm lâm sàng9).
Ứng viên liệu pháp chỉnh sửa gen EDIT-101 sử dụng CRISPR/Cas9 đã được báo cáo trong phát triển tiền lâm sàng cho LCA10 (biến thể CEP290)11).
Xét nghiệm di truyền tiền làm tổ (PGT-M) có thể được thực hiện cho các bệnh di truyền trội và lặn trên nhiễm sắc thể thường, và có thể được xem xét trong khuôn khổ đạo đức12).
Giải trình tự toàn bộ hệ gen (WGS) đã cho thấy có thể làm tăng tỷ lệ chẩn đoán phân tử trong các bệnh võng mạc di truyền so với các xét nghiệm di truyền tiêu chuẩn hiện có13).
Các công cụ dự đoán tính gây bệnh của biến thể gen bằng trí tuệ nhân tạo (AI) đang được phát triển, và hy vọng sẽ cải thiện độ chính xác trong diễn giải biến thể.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.