مشاورهٔ ژنتیک یک خدمت پزشکی است که هدف آن «ارائهٔ اطلاعات ژنتیکی درست» است. این خدمت به بیماران و خانوادهها دربارهٔ تشخیص، الگوی وراثت، خطر تکرار، آزمایشهای موجود و روشهای درمان بیماریهای ژنتیکی اطلاعات میدهد و از تصمیمگیری مستقل حمایت میکند. این تعریف در سطح بینالمللی نیز پذیرفته شده است و سه ستون مشاورهٔ ژنتیک عبارتاند از «ارائهٔ اطلاعات»، «حمایت روانی» و «حمایت در تصمیمگیری»3).
بیماریهای چشمی ارثی حدود ۴۳٪ از اختلال بینایی مادرزادی را تشکیل میدهند1). فراوانی ناهنجاریهای کروموزومی در فرزندان حدود ۰٫۵ تا ۱٪ است. اختلال دید رنگی قرمز-سبز یکی از شایعترین بیماریهای چشمی ارثی است و در حدود ۵٪ مردان و ۰٫۲٪ زنان دیده میشود. شیوع رتینیت پیگمانته حدود ۱/۴٬۰۰۰ تا ۱/۵٬۰۰۰ است و یکی از علل اصلی اختلال بینایی به شمار میرود2).
دامنهٔ مشاورهٔ ژنتیک فقط بیمار مبتلا به بیماری ارثی را شامل نمیشود، بلکه اعضای خانوادهای را که ممکن است به آن مبتلا شوند و نیز ناقلانی را که نگران انتقال آن به فرزندان آیندهٔ خود هستند دربر میگیرد. چشمپزشک در کنار انجام تشخیص، با متخصصان ژنتیک و مشاوران ژنتیک دارای گواهی همکاری میکند تا اطلاعات لازم را ارائه دهد.
Qمشاورهٔ ژنتیک را از کجا میتوان دریافت کرد؟
A
این خدمت را میتوان در بخشهای ژنتیک بیمارستانهای دانشگاهی یا بیمارستانهای مرجع، یا در مراکزی که مشاور ژنتیک دارای گواهی دارند دریافت کرد. با مراجعه به فهرست مشاوران ژنتیک دارای گواهیِ انجمن مشاورهٔ ژنتیک ژاپن و انجمن ژنتیک انسانی ژاپن میتوان این مراکز را پیدا کرد. وبسایت مرکز اطلاعات بیماریهای نادر نیز راهنمای محلهای مشاوره را ارائه میدهد.
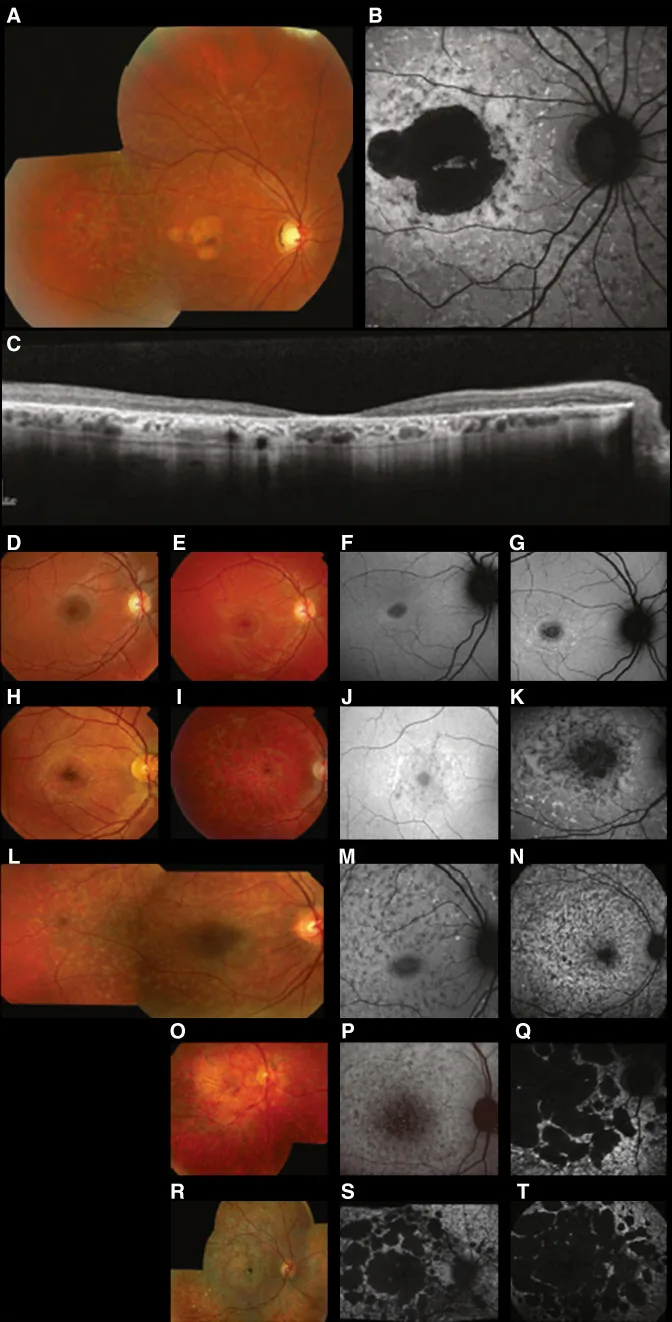
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
تصاویر چندوجهیِ تیپیکِ بیماری استارگاردت (STGD1): عکس رنگی فوندوس لکههای زرد-سفید را در سطح اپیتلیوم رنگدانهای شبکیه و آتروفی ماکولا نشان میدهد (A)؛ اتوفلورسانس فوندوس یک ناحیه کمفلورسانس در ماکولا همراه با فلورسانس غیرطبیعی اطراف آن را نشان میدهد (B)؛ و SD-OCT از دست رفتن واضح لایههای خارجی شبکیه و RPE را بههمراه کانونهای هایپررفلکتیو متناظر با لکهها نشان میدهد (C). این با بیماری استارگاردت (یک دیستروفی شبکیهای اتوزومال مغلوب ناشی از جهشهای ژن ABCA4) که در بخش «2. الگوهای وراثت و بیماریهای مهم چشمیِ ارثی» مطرح شده است، مطابقت دارد.
الگوی وراثت از اطلاعات اصلی در مشاوره ژنتیک است و به چهار الگوی اصلی تقسیم میشود.
وراثت اتوزومال غالب
شرط بروز: بیماری با جهش در یک آلل (حالت هتروزیگوت) ایجاد میشود.
ویژگیهای شجرهنامه: افراد مبتلا در نسلهای پیاپی دیده میشوند.
خطر تکرار: احتمال انتقال به فرزند 50% است.
نکته: اگر نفوذپذیری 100% نباشد، ممکن است یک نسل رد شود.
وراثت اتوزومال مغلوب
شرط بروز: بیماری زمانی ایجاد میشود که هر دو آلل جهشیافته باشند (هموزیگوت یا هتروزیگوت مرکب).
ویژگیهای شجرهنامه: افراد مبتلا در میان خواهر و برادرها دیده میشوند. والدین معمولاً ناقل (هتروزیگوت) هستند.
خطر تکرار: احتمال بیماری در فرزندِ دو ناقل 25% است.
روند اخیر: با کمتر شدن ازدواج بین پسرعموها و دخترعموها، نسبت هتروزیگوتهای مرکب افزایش یافته است.
وراثت وابسته به X
شرط بروز: بیشتر بیماران مرد هستند (همیزیگوت).
در زنان: چون دو کروموزوم X دارند، اگر یکی دچار جهش شود، فرد ناقل میشود.
ویژگیهای شجرهنامه: مبتلایان بیشتر مرد هستند و انتقال از مادر به پسر انجام میشود.
خطر عود: احتمال ابتلای پسرِ مادرِ ناقل 50٪ است.
وراثت مادری (وراثت میتوکندریایی)
ویژگی: DNA میتوکندریایی اسپرم (mtDNA) هنگام لقاح تقریباً بهطور کامل تجزیه میشود. بنابراین فقط از مادر به فرزند منتقل میشود.
Qاگر یکی از والدین دچار بیماری چشمی ارثی باشد، احتمال انتقال آن به فرزند چقدر است؟
A
الگوی وراثت متفاوت است. در وراثت اتوزومال غالب، احتمال انتقال از والد مبتلا به فرزند 50٪ است. در وراثت اتوزومال مغلوب، اگر هر دو والد ناقل باشند، احتمال ابتلای فرزند 25٪ است. در وراثت وابسته به کروموزوم X، احتمال ابتلای پسرِ مادرِ ناقل 50٪ است و در وراثت مادری (میتوکندریایی)، اگر مادر مبتلا یا ناقل باشد، ممکن است به همه فرزندان منتقل شود. برای شرایط فردی، مشاوره با متخصص ژنتیک یا مشاور ژنتیک دارای گواهی توصیه میشود.
سازوکارهای بروز بیماریهای چشمی ارثی بر اساس نوع اثری که جهش ژنتیکی بر عملکرد پروتئین میگذارد، طبقهبندی میشوند.
هاپلوانسافیسیانسی (haploinsufficiency): با از دست رفتن عملکرد تنها یک آلل، فنوتیپ ظاهر میشود. نمونهٔ شاخص آن آنریدی مادرزادی (جهش PAX6) است. یک آلل سالم بهتنهایی نمیتواند مقدار محصول ژنی لازم برای تشکیل طبیعی بافتها را فراهم کند.
اثر غالب منفی (dominant negative effect): پروتئین جهشیافته، عملکرد پروتئین طبیعی را بهصورت رقابتی و ساختاری مهار میکند. در سندرم مارفان (جهش FBN1)، مولکولهای جهشیافتهٔ فیبریلین-1 تشکیل ماتریکس خارجسلولی را مختل میکنند.
جهشهای میتوکندریایی: در نوروپاتی بینایی ارثی لِبِر (LHON)، سه جهش نقطهای 11778، 3460 و 14484 حدود 90٪ از همهٔ جهشها را تشکیل میدهند. اختلال در تولید انرژی به سلولهای گانگلیونی شبکیه آسیب میزند.
جهشهای دِ نوو: جهشهایی که در والدین وجود ندارند و هنگام تشکیل تخمک یا اسپرم بهطور جدید ایجاد میشوند. نمونهٔ شاخص آن آماوروز مادرزادی لِبِر ناشی از جهشهای CRX است. این جهشها میتوانند بهصورت موارد تکگیر نیز دیده شوند، حتی وقتی هیچ فرد دیگری در شجرهنامه مبتلا نباشد.
هتروزیگوت مرکب: در بیماریهای اتوزومال مغلوب، حالتی است که در آن دو جهش متفاوت در دو آلل رخ میدهد. با کاهش اخیر ازدواج فامیلیِ پسرعمو/دخترعموها، نسبت هتروزیگوتهای مرکب در مقایسه با هموزیگوتها افزایش یافته است.
دیسومی یکوالدی: پدیدهای که در آن هر دو نسخهٔ یک کروموزوم از یک والد به ارث میرسند و کروموزومِ والد دیگر وجود ندارد. این حالت میتواند بهصورت یک مورد ظاهراً پراکنده10) ظاهر شود.
بیش از 100 ژنِ عامل برای رتینیت پیگمنتوزا شناسایی شدهاند، و حتی یک فنوتیپِ یکسان نیز میتواند توسط ژنهای متفاوت زیادی ایجاد شود2). این تنوع ژنتیکی تشخیص را دشوار میکند.
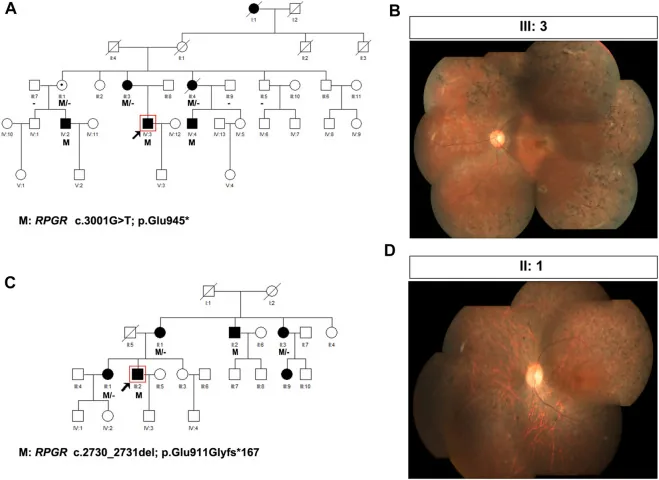
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
شجرهنامهها و عکسهای فوندوسِ دو خانواده مبتلا به رتینیت پیگمانتوزای وابسته به X (واریانتهای RPGR: c.3001G>T و c.2730_2731del). در شجرهنامهها از نمادهای استاندارد (مربعهای پر برای مردان مبتلا، دایرههای نقطهچین برای زنان ناقل) برای نشان دادن الگوی وراثت استفاده شده است، و عکسهای فوندوسِ مربوطه رنگدانهگذاری پیشرفته به شکل خارهای استخوانی و آتروفی شبکیه را نشان میدهند. این با رسم شجرهنامه و تعیین الگوی وراثت که در بخش «4. مشاوره ژنتیک عملی و آزمایش ژنتیک» مطرح شده است، مطابقت دارد.
برای انجام درست مشاوره ژنتیک، آمادهسازی زیر لازم است.
تهیه شجرهنامه (pedigree): گرفتن شرححال خانوادگی دستکم سه نسل و ثبت آن به صورت شجرهنامه. پراکندگی عمودی و افقی افراد مبتلا میتواند به حدس الگوی وراثت کمک کند.
حدس زدن الگوی وراثت: بر اساس شجرهنامه و فنوتیپ، مشخص کنید که آیا وراثت اتوزومی غالب، اتوزومی مغلوب، وابسته به X یا مادری است.
توضیح آزمایش ژنتیک و گرفتن رضایت کتبی: پیش از بررسی اطلاعات ژنتیکی بیمار، لازم است معنی و مشکلات آزمایش بهطور کامل توضیح داده شود و رضایت کتبی گرفته شود.
ناشناسسازی نتایج آزمایش DNA و مدیریت اطلاعات: با ناشناسسازی قابل اتصال از حریم خصوصی محافظت شود.
تعیین از پیش نحوه برخورد با یافتههای اتفاقی: پیش از آزمایش، با بیمار درباره سیاست اطلاعرسانی در صورت یافتن یافتههای اتفاقی جدی و تهدیدکننده حیات توافق شود.
بررسی همزمان ژنهای مرتبط با بیماریهای شبکیه. میزان تشخیص بالا5)
تحلیل اگزوم (NGS)
تمام نواحی اگزونی
شناسایی واریانتهای ناشناخته. وقتی ژنهای بیماریزا زیاد باشند، مفید است
تحلیل کل ژنوم (WGS)
کل ژنوم
ممکن است نرخ تشخیص بالاتری نسبت به آزمایشهای NGS هدفمند موجود داشته باشد13)
در دیستروفی ارثی شبکیه، هنگامی که به بیماری ناشی از جهش ژن RPE65 مشکوک باشیم، و در مواردی مانند کمک به تعیین صلاحیت برای ژندرمانی، برخی از آزمایشهایی که شرایط را دارند میتوانند بهعنوان خدمات درمانی تحت پوشش بیمه انجام شوند4).
تفسیر واریانتها نیازمند داوری دقیق است. شناخته شده است که حدود 30٪ از توضیحاتی که در مقالات بهعنوان «واریانت» منتشر میشوند، در واقع پلیمورفیسم (واریانتهای طبیعی) هستند. بهطور کلی، تغییرات توالی باز که در 1 نفر یا بیشتر از هر 100 فرد سالم دیده میشوند، باید بهعنوان پلیمورفیسم در نظر گرفته شوند.
از پایگاههای داده اصلی زیر استفاده میشود.
OMIM (Online Mendelian Inheritance in Man): پایگاه داده جامع بیماریها و ژنهای ارثی
GeneReviews: اطلاعات مشاوره ژنتیک را برای هر بیماری ارائه میکند
RetNet (Retinal Information Network): پایگاه داده ژنی تخصصی در بیماریهای شبکیه
Qآیا هزینه آزمایش ژنتیک تحت پوشش بیمه است؟
A
در برخی بیماریهای چشمی ارثی، آزمایش ژنتیک میتواند بهعنوان خدمات درمانی تحت پوشش بیمه انجام شود، زمانی که اندیکاسیون درمان مشخص شده و شرایط مرکز تعیینشده برآورده شود. با این حال، اینکه تحت پوشش باشد یا نه به نوع آزمایش و بیماری مورد نظر بستگی دارد، بنابراین باید در مرکز مراجعهشده آن را بررسی کرد. آزمایشهایی که تحت پوشش بیمه نیستند ممکن است بهصورت آزاد محاسبه شوند. در بیماریهای مشخصشده و سختدرمان (مانند رتینیت پیگمانتوزا)، ممکن است حمایت مالی از طریق نظام یارانه هزینههای درمانی بیماریهای مشخصشده و سختدرمان نیز در دسترس باشد4).
در اجرای مشاوره ژنتیک، همکاری با پزشک متخصص ژنتیک و مشاور ژنتیک دارای گواهی ایدهآل شمرده میشود. در بیمارستانهای دانشگاهی ممکن است بررسی توسط کمیته اخلاق لازم باشد. از آنجا که اطلاعات ژنتیک میتواند نهتنها بر خود بیمار بلکه بر اعضای خانواده نیز اثر بگذارد، در نحوه برخورد با آن دقت ویژهای لازم است. بحثهای اجتماعی درباره پیشگیری از تبعیض ناعادلانه بر پایه اطلاعات ژنتیک نیز در حال پیشرفت است8).
در بیماران مبتلا به بیماریهای نادر، سهم پرداختی از هزینههای پزشکی کاهش مییابد. برای مجموع هزینههای سرپایی، بستری و دارو، سقف ماهانه تعیین شده و دستهبندی بر اساس درآمد اعمال میشود.
داروی درمان ژنی voretigene neparvovec برای دیستروفی ارثی شبکیه ناشی از جهشهای ژن RPE65، یک فرآورده برای تزریق زیرشبکیهای با استفاده از ناقل AAV2 است. اثربخشی و ایمنی آن در کارآزماییهای تصادفیسازیشده و کنترلشده تأیید شده است6). تصمیمگیری درباره مناسب بودن درمان، مستلزم تأیید نوع بیماری از طریق آزمایش ژنتیکی است.
در مورد درمان با الیگونوکلئوتیدهای آنتیسنس (ASO)، تزریق داخل زجاجیهای یک فرآورده برای آموروز مادرزادی لبر نوع 10 (LCA10) ناشی از جهشهای CEP290 در کارآزماییهای بالینی در حال بررسی است9).
پیوند خودی سلولهای اپیتلیوم رنگدانهای شبکیه با استفاده از سلولهای iPS بهعنوان پزشکی بازساختی برای دژنراسیون ماکولای وابسته به سن در حال بررسی است7). اگرچه این حوزهای متفاوت از بیماریهای ارثی شبکیه است، اما بهعنوان نمونهای واقعی از درمان سلولی شبکیه مورد توجه قرار گرفته است.
Qآیا ژندرمانی اکنون در دسترس است؟
A
در دیستروفی شبکیه ارثی ناشی از جهش RPE65، اثربخشی و ایمنی voretigene neparvovec در RCTها6) نشان داده شده است. برای تأیید مناسب بودن درمان، تعیین نوع بیماری با آزمایش ژنتیک لازم است. در LCA10 همراه با جهش CEP290، تزریق درونزجاجیهای فرآوردههای ASO در کارآزماییهای بالینی در حال بررسی است9).
وراثت اتوزومال غالب، اتوزومال مغلوب و وابسته به X از قوانین مندل پیروی میکند. با این حال، عوامل زیر میتوانند پیشبینی ساده را دشوار کنند.
نفوذپذیری (penetrance): حتی اگر فرد جهش را داشته باشد، همه به بیماری مبتلا نمیشوند. وقتی نفوذپذیری پایین باشد، صفت ممکن است از یک نسل بگذرد و از روی شجرهنامه تعیین الگوی وراثت دشوار میشود.
بروزپذیری (expressivity): حتی در میان اعضای خانوادهای که همان ژن جهشیافته را دارند، شدت علائم میتواند متفاوت باشد.
جهش افزایش عملکرد (gain-of-function): سازوکاری که در آن پروتئین جهشیافته عملکرد زیانبار جدیدی به دست میآورد. این با اثر غالب-منفی معمول متفاوت است.
میتوکندریها در سیتوپلاسم قرار دارند و تنها mtDNA که از تخمک مادر منشأ میگیرد به فرزند منتقل میشود. در هر سلول هزاران نسخه mtDNA وجود دارد و ممکن است حالتی رخ دهد که mtDNA جهشیافته و mtDNA طبیعی با هم وجود داشته باشند (heteroplasmy). هرچه نسبت heteroplasmy بیشتر باشد، علائم معمولاً شدیدتر است. در نوروپاتی بینایی ارثی لبر (LHON)، سه جهش—11778 (شایعترین)، 3460 و 14484—حدود 90٪ از همه جهشها را تشکیل میدهند.
دیسومی یکوالدی حالتی است که در آن هر دو کروموزوم یک جفت از یک والد به ارث میرسند و کروموزومی از والد دیگر وجود ندارد. فرزندی که از یک ناقل بیماری اتوزومال مغلوب به دنیا میآید، حتی اگر والد دیگر ناقل نباشد، ممکن است بیمار شود و در ظاهر بهصورت یک مورد پراکنده دیده شود10). همراه با جهشهای de novo و هتروزیگوتهای مرکب، باید بیماری ارثی را حتی در نبود سابقه خانوادگی در نظر گرفت.
در LCA10 همراه با جهشهای CEP290، اصلاح splicing با درمان ASO در کارآزماییهای بالینی در حال مطالعه است9).
نامزد درمان ویرایش ژن EDIT-101 با استفاده از CRISPR/Cas9 برای LCA10 (جهشهای CEP290) در مرحله توسعه پیشبالینی گزارش شده است11).
آزمایش ژنتیک پیش از لانهگزینی (PGT-M) برای بیماریهای ارثی اتوزومال غالب و مغلوب قابل انجام است و ممکن است در چارچوبهای اخلاقی مورد بررسی قرار گیرد12).
نشان داده شده است که تعیین توالی کل ژنوم (WGS) میتواند نرخ تشخیص مولکولی را در بیماریهای ارثی شبکیه نسبت به آزمایشهای ژنتیکی استاندارد موجود افزایش دهد13).
ابزارهای پیشبینی بیماریزایی واریانتهای ژنی با استفاده از هوش مصنوعی (AI) در حال توسعه هستند و انتظار میرود دقت تفسیر واریانتها بهبود یابد.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.