การให้คำปรึกษาทางพันธุกรรม เป็นบริการทางการแพทย์ที่ให้ข้อมูลทางพันธุกรรมที่ถูกต้องและช่วยให้ผู้ป่วยและครอบครัวตัดสินใจได้ด้วยตนเองรูปแบบการถ่ายทอดมี 4 แบบ ได้แก่ แบบยีนเด่นบนโครโมโซมร่างกาย แบบยีนด้อยบนโครโมโซมร่างกาย แบบเชื่อมโยงกับโครโมโซม X และแบบถ่ายทอดทางมารดา (ไมโตคอนเดรีย) และความเสี่ยงการกลับเป็นซ้ำแตกต่างกัน
โรคตาทางพันธุกรรม คิดเป็นประมาณ 43% ของความบกพร่องทางการมองเห็น แต่กำเนิด และความชุกของ retinitis pigmentosa อยู่ที่ประมาณ 1/4,000 ถึง 1/5,0002) .ต้องได้รับความยินยอมเป็นลายลักษณ์อักษรก่อนการตรวจทางพันธุกรรม (วิธี PCR, วิธี Sanger และ NGS)
ประมาณ 30% ของคำอธิบายที่ระบุว่าเป็นการกลายพันธุ์ในบทความ แท้จริงแล้วเป็นพอลิมอร์ฟิซึม ดังนั้นการตีความการกลายพันธุ์จึงต้องใช้การพิจารณาอย่างรอบคอบ
ในโรคจอประสาทตา เสื่อมทางพันธุกรรมที่เกิดจากการกลายพันธุ์ของ RPE65 ประสิทธิผลและความปลอดภัยของยาบำบัดยีน voretigene neparvovec ได้แสดงให้เห็นแล้วในการทดลองแบบสุ่มมีกลุ่มควบคุม (RCT)6) .
โรคเรตินิติสพิกเมนโตซา ต้อหิน แต่กำเนิด และโรคกระจกตา เสื่อมแบบหยดเจลาตินเป็นโรคที่กำหนดให้เป็นโรครักษายาก และมีสิทธิ์ได้รับเงินช่วยเหลือค่ารักษาพยาบาล.
การให้คำปรึกษาทางพันธุกรรม เป็นบริการทางการแพทย์ที่มีเป้าหมายเพื่อ «ให้ข้อมูลทางพันธุกรรมที่ถูกต้อง» โดยให้ข้อมูลแก่ผู้ป่วยและครอบครัวเกี่ยวกับการวินิจฉัย รูปแบบการถ่ายทอด ความเสี่ยงที่จะเกิดซ้ำ การตรวจที่มีอยู่ และวิธีการรักษาของโรคทางพันธุกรรม พร้อมทั้งสนับสนุนการตัดสินใจอย่างเป็นอิสระ คำจำกัดความนี้ได้รับการยอมรับในระดับนานาชาติด้วย และเสาหลักสามประการของการให้คำปรึกษาทางพันธุกรรม คือ «การให้ข้อมูล» «การสนับสนุนทางจิตใจ» และ «การสนับสนุนการตัดสินใจ»3) .
โรคตาทางพันธุกรรม คิดเป็นประมาณ 43% ของความบกพร่องทางการมองเห็น แต่กำเนิด1) . ความถี่ของความผิดปกติของโครโมโซมในบุตรอยู่ที่ประมาณ 0.5-1%. ภาวะบกพร่องการมองเห็น สีแดง-เขียวเป็นหนึ่งในโรคตาทางพันธุกรรม ที่พบบ่อยที่สุด พบในผู้ชายประมาณ 5% และผู้หญิงประมาณ 0.2%. ความชุกของโรคเรตินิติสพิกเมนโตซาอยู่ที่ประมาณ 1/4,000-1/5,000 และเป็นหนึ่งในสาเหตุหลักของความบกพร่องทางการมองเห็น 2) .
ผู้ที่อยู่ในขอบเขตของการให้คำปรึกษาทางพันธุกรรม ไม่ได้มีเพียงผู้ป่วยโรคทางพันธุกรรมเท่านั้น แต่ยังรวมถึงสมาชิกในครอบครัวที่อาจเป็นโรคได้ และผู้เป็นพาหะที่กังวลว่าจะถ่ายทอดไปยังบุตรในอนาคต จักษุแพทย์มีบทบาทในการวินิจฉัย และทำงานร่วมกับแพทย์ผู้เชี่ยวชาญด้านพันธุศาสตร์และนักให้คำปรึกษาทางพันธุกรรมที่ได้รับการรับรองเพื่อให้ข้อมูล
Q
สามารถรับการให้คำปรึกษาทางพันธุกรรมได้ที่ไหน?
A
สามารถรับได้ที่แผนกเวชพันธุศาสตร์ของโรงพยาบาลมหาวิทยาลัยหรือโรงพยาบาลศูนย์ หรือในสถานพยาบาลที่มีนักให้คำปรึกษาทางพันธุกรรมที่ได้รับการรับรอง สามารถค้นหาสถานพยาบาลได้จากรายชื่อนักให้คำปรึกษาทางพันธุกรรมที่ได้รับการรับรองของสมาคมให้คำปรึกษาทางพันธุกรรมแห่งประเทศญี่ปุ่นและสมาคมพันธุศาสตร์มนุษย์แห่งประเทศญี่ปุ่น เว็บไซต์ของศูนย์ข้อมูลโรคหายากยังมีข้อมูลแนะนำสถานที่ปรึกษา
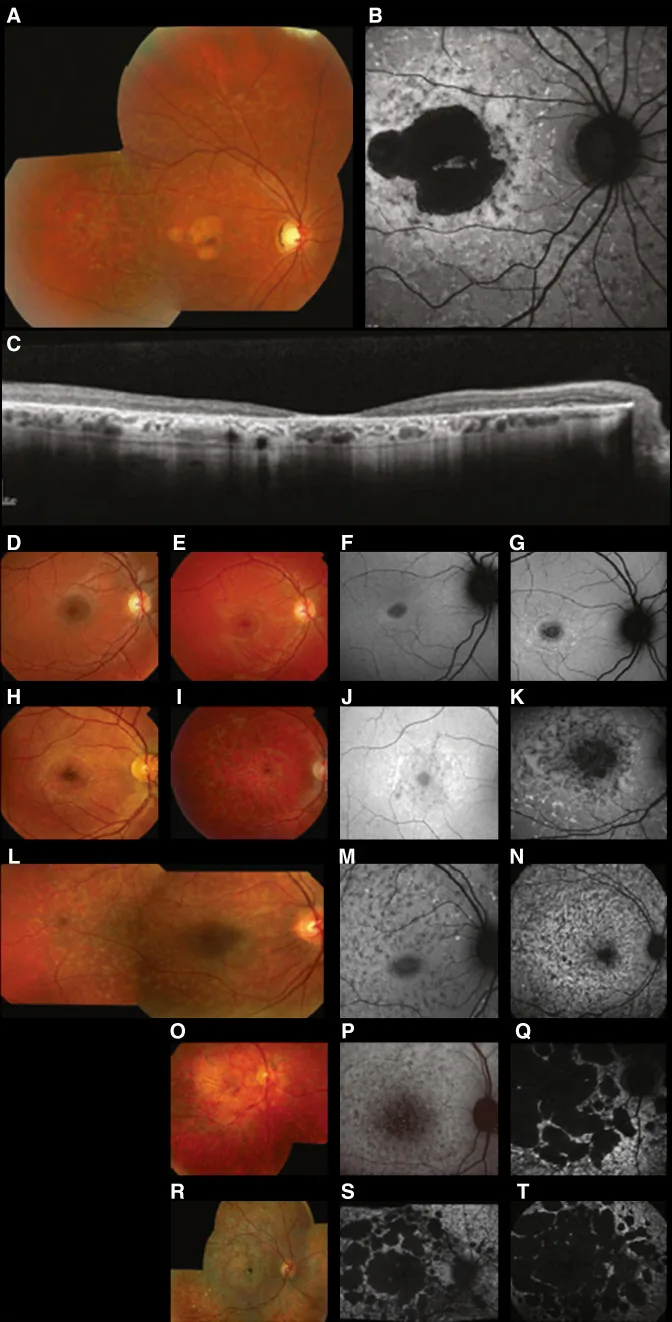
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
ภาพหลายรูปแบบแบบฉบับของ
โรคสตาร์การ์ดต์ (STGD1): ภาพถ่ายฟันดัสแบบสีแสดงจุดสีเหลืองขาวที่ระดับเยื่อบุผิวสีของ
จอประสาทตา และภาวะฝ่อของ
จุดภาพชัด (A); ภาพฟันดัสออโตฟลูออเรสเซนซ์แสดงบริเวณฟลูออเรสเซนซ์ต่ำที่
จุดภาพชัด พร้อมฟลูออเรสเซนซ์ผิดปกติรอบ ๆ (B); และ SD-
OCT แสดงการสูญเสียอย่างชัดเจนของชั้น
จอประสาทตา ส่วนนอกและ
RPE พร้อมจุดสะท้อนสูงที่สอดคล้องกับจุดดังกล่าว (C). สอดคล้องกับ
โรคสตาร์การ์ดต์ (โรค
จอประสาทตา เสื่อมแบบยีนด้อยบนโครโมโซมร่างกายจากการกลายพันธุ์ของยีน ABCA4) ที่กล่าวถึงในหัวข้อ “2. รูปแบบการถ่ายทอดทางพันธุกรรมและ
โรคตาทางพันธุกรรม ที่สำคัญ”.
รูปแบบการถ่ายทอดทางพันธุกรรมเป็นข้อมูลสำคัญในการให้คำปรึกษาทางพันธุกรรม และแบ่งออกเป็น 4 รูปแบบหลัก.
การถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่น
เงื่อนไขการเกิดโรค : โรคเกิดขึ้นเมื่อมียีนแอลลีลใดแอลลีลหนึ่งเกิดการกลายพันธุ์ (ภาวะเฮเทอโรไซกัส).
ลักษณะผังเครือญาติ : ผู้ป่วยจะปรากฏต่อเนื่องในแต่ละรุ่น.
ความเสี่ยงการเกิดซ้ำ : โอกาสถ่ายทอดไปยังบุตรคือ 50%.
ข้อควรระวัง : หากการแทรกซึมของยีนไม่ถึง 100% อาจดูเหมือนข้ามรุ่นได้.
การถ่ายทอดทางพันธุกรรมแบบออโตโซมด้อย
เงื่อนไขการเกิดโรค : โรคเกิดขึ้นเมื่อแอลลีลทั้งสองเกิดการกลายพันธุ์ (โฮโมไซกัสหรือเฮเทอโรไซกัสแบบคู่ผสม).
ลักษณะผังเครือญาติ : ผู้ป่วยจะพบในหมู่พี่น้อง พ่อแม่มักเป็นพาหะ (เฮเทอโรไซกัส).
ความเสี่ยงการเกิดซ้ำ : โอกาสเกิดโรคในบุตรของพาหะสองคนคือ 25%.
แนวโน้มในปัจจุบัน : เนื่องจากการแต่งงานระหว่างลูกพี่ลูกน้องลดลง สัดส่วนของผู้ที่เป็นเฮเทอโรไซกัสแบบคู่ผสมจึงเพิ่มขึ้น.
การถ่ายทอดทางโครโมโซม X
เงื่อนไขการเกิดโรค : ผู้ป่วยส่วนใหญ่เป็นเพศชาย (เฮมีไซโกต).
ในผู้หญิง : เนื่องจากมีโครโมโซม X สองแท่ง หากมีการกลายพันธุ์ในหนึ่งแท่งจะกลายเป็นพาหะ.
ลักษณะในผังครอบครัว : ผู้ป่วยมักเป็นเพศชายเป็นส่วนใหญ่ และถ่ายทอดจากแม่สู่ลูกชาย.
ความเสี่ยงในการเกิดซ้ำ : โอกาสที่บุตรชายของมารดาที่เป็นพาหะจะเป็นโรคคือ 50%.
การถ่ายทอดทางมารดา (การถ่ายทอดทางไมโทคอนเดรีย)
ลักษณะ : ดีเอ็นเอไมโทคอนเดรีย (mtDNA) ของอสุจิจะถูกสลายเกือบทั้งหมดเมื่อเกิดการปฏิสนธิ ดังนั้นจึงถ่ายทอดจากแม่ไปสู่ลูกเท่านั้น.
โรคตัวอย่าง : โรคเส้นประสาทตา เสื่อมทางพันธุกรรมของ Leber (LHON ).
จุดแยกจากการถ่ายทอดแบบเชื่อมโยงกับโครโมโซม X : ต่างกันตรงที่พบผู้ป่วยหญิงได้ด้วย.
เฮเทอโรพลาสมี : เมื่อ mtDNA ปกติและ mtDNA กลายพันธุ์อยู่ร่วมกัน ลักษณะอาการจะแตกต่างกันได้.
ตำแหน่ง ชื่อโรค รูปแบบการถ่ายทอด ยีนที่ก่อโรค กระจกตา ภาวะกระจกตา เสื่อมชนิดหยดเจลาติน ยีนด้อยแบบออโตโซม TACSTD2 (1p) กระจกตา ภาวะกระจกตา เสื่อมชนิดเม็ดเล็ก ชนิดร่างแห และชนิดอะเวลลิโน ยีนเด่นแบบออโตโซม TGFBI (5q) เลนส์ตา ต้อกระจกแต่กำเนิด ยีนเด่นแบบออโตโซม (หลายยีน) — เลนส์ กลุ่มอาการมาร์แฟน เด่นแบบออโตโซม FBN1 (15q) จอประสาทตา โรคจอประสาทตา เสื่อมจากเม็ดสี เด่นแบบออโตโซม, ด้อยแบบออโตโซม และเชื่อมโยงกับโครโมโซม X ยีนก่อโรคมากกว่า 100 ยีน2) จอประสาทตา โรคสตาร์การ์ดต์ ด้อยแบบออโตโซม ABCA4 (1p) จอประสาทตา ภาวะเรติโนสคีซิสในวัยเยาว์ เชื่อมโยงกับโครโมโซม X RS1 (Xp) จอประสาทตา เรติโนบลาสโตมา เด่นแบบออโตโซม RB1 (13q) เส้นประสาทตา โรคเส้นประสาทตา เสื่อมทางพันธุกรรมของเลเบอร์ถ่ายทอดทางมารดา (mtDNA) — เส้นประสาทตา โรคฝ่อของเส้นประสาทตา แบบเด่นบนโครโมโซมร่างกาย เด่นบนโครโมโซมร่างกาย OPA1 (3q) การมองเห็น สีภาวะบกพร่องการมองเห็น สีแดง-เขียว เชื่อมโยงกับโครโมโซม X —
Q
หากพ่อหรือแม่เป็นโรคตาทางพันธุกรรม โอกาสถ่ายทอดไปยังลูกมีเท่าไร?
A
รูปแบบการถ่ายทอดทางพันธุกรรมแตกต่างกัน ในการถ่ายทอดแบบเด่นบนโครโมโซมร่างกาย โอกาสที่พ่อหรือแม่ที่เป็นโรคจะถ่ายทอดไปยังลูกคือ 50% ในการถ่ายทอดแบบด้อยบนโครโมโซมร่างกาย หากพ่อและแม่เป็นพาหะทั้งคู่ โอกาสที่ลูกจะเป็นโรคคือ 25% ในการถ่ายทอดแบบเชื่อมโยงกับโครโมโซม X โอกาสที่ลูกชายของแม่ที่เป็นพาหะจะเป็นโรคคือ 50% และในการถ่ายทอดทางสายมารดา (ไมโทคอนเดรีย) หากแม่เป็นโรคหรือเป็นพาหะ โรคอาจถ่ายทอดไปยังลูกทุกคนได้ สำหรับแต่ละกรณี แนะนำให้ปรึกษาแพทย์ผู้เชี่ยวชาญด้านพันธุศาสตร์หรือที่ปรึกษาทางพันธุกรรมที่ได้รับการรับรอง
กลไกการเกิดโรคตาทางพันธุกรรม แบ่งตามชนิดของผลที่การกลายพันธุ์ของยีนมีต่อการทำงานของโปรตีน
ภาวะฮาพโลอินซัฟฟิเชียนซี (haploinsufficiency) : ฟีโนไทป์เกิดขึ้นเมื่อสูญเสียการทำงานของอัลลีลเพียงหนึ่งข้าง ตัวอย่างที่พบบ่อยคือภาวะม่านตา ไม่มีแต่กำเนิด (การกลายพันธุ์ของ PAX6) อัลลีลปกติเพียงหนึ่งตัวไม่สามารถให้ผลิตภัณฑ์ยีนได้เพียงพอสำหรับการสร้างเนื้อเยื่อตามปกติ.ผลเด่นเชิงลบ (dominant negative effect) : โปรตีนที่กลายพันธุ์ยับยั้งการทำงานของโปรตีนปกติทั้งแบบแข่งขันและแบบโครงสร้าง ในกลุ่มอาการมาร์แฟน (การกลายพันธุ์ของ FBN1) โมเลกุล fibrillin-1 ที่กลายพันธุ์รบกวนการสร้างเมทริกซ์นอกเซลล์.การกลายพันธุ์ของไมโทคอนเดรีย : ในโรคประสาทตาเสื่อมทางพันธุกรรมของเลเบอร์ (LHON ) การกลายพันธุ์แบบจุด 3 ตำแหน่ง ได้แก่ 11778, 3460 และ 14484 คิดเป็นประมาณ 90% ของการกลายพันธุ์ทั้งหมด ความบกพร่องในการสร้างพลังงานทำลายเซลล์ปมประสาทของจอประสาทตา .การกลายพันธุ์ de novo : การกลายพันธุ์ที่ไม่ได้มีอยู่ในพ่อแม่และเกิดขึ้นใหม่ระหว่างการสร้างไข่หรืออสุจิ ตัวอย่างที่สำคัญคือภาวะตาบอดแต่กำเนิดของเลเบอร์ที่เกิดจากการกลายพันธุ์ของ CRX นอกจากนี้ยังอาจพบเป็นรายเดี่ยวได้เมื่อไม่มีใครในผังเครือญาติที่ได้รับผลกระทบ.เฮเทอโรไซโกตแบบผสม (compound heterozygote) : ในโรคยีนด้อยบนโครโมโซมร่างกาย เป็นรูปแบบที่อัลลีลทั้งสองมีการกลายพันธุ์ต่างกันคนละแบบ เมื่อการแต่งงานในเครือญาติลดลงในช่วงหลัง สัดส่วนของเฮเทอโรไซโกตแบบผสมจึงเพิ่มขึ้นเมื่อเทียบกับโฮโมไซโกต.ยูนิโพเรนทัลไดโซมี : ภาวะที่โครโมโซมคู่เดียวกันทั้งสองสำเนาได้รับมาจากพ่อหรือแม่เพียงฝ่ายเดียว ขณะที่โครโมโซมจากอีกฝ่ายหายไป อาจแสดงเป็นกรณีที่ดูเหมือนเกิดขึ้นแบบประปราย10) .
มีการระบุยีนก่อโรคของภาวะจอประสาทตา เสื่อมสีชนิดเรตินิติสพิกเมนโตซามากกว่า 100 ยีน และแม้แต่ฟีโนไทป์เดียวกันก็อาจเกิดจากยีนที่ต่างกันได้หลายชนิด2) . ความหลากหลายทางพันธุกรรมนี้ทำให้การวินิจฉัยยากขึ้น.
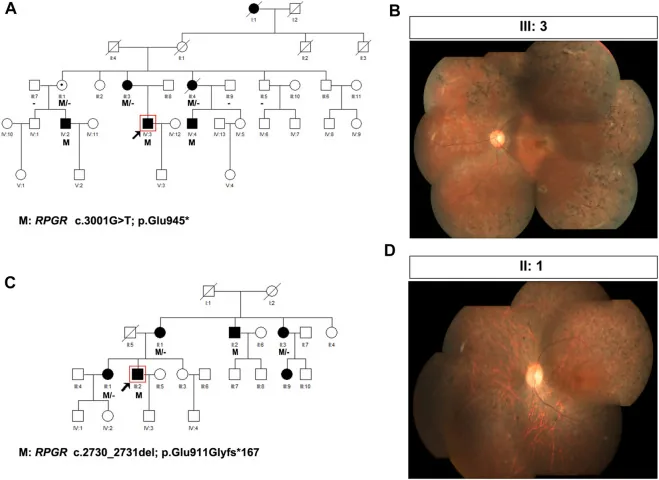
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
แผนผังเครือญาติและภาพถ่ายก้นตาของสองครอบครัวที่มีโรค
จอประสาทตา เสื่อมสีชนิดเชื่อมโยงกับโครโมโซม X (ความแปรผันของ RPGR: c.3001G>T และ c.2730_2731del) แผนผังเครือญาติใช้สัญลักษณ์มาตรฐาน (สี่เหลี่ยมทึบสำหรับผู้ชายที่เป็นโรค วงกลมเส้นประสำหรับผู้หญิงพาหะ) เพื่อแสดงรูปแบบการถ่ายทอด และภาพถ่ายก้นตาที่สอดคล้องกันแสดงการสะสมสีแบบก้างปลาที่ลุกลามและจอตาฝ่อ ข้อความนี้สอดคล้องกับการจัดทำแผนผังเครือญาติและการระบุรูปแบบการถ่ายทอดที่กล่าวถึงในหัวข้อ “4.
การให้คำปรึกษาทางพันธุกรรม และการตรวจยีนในทางปฏิบัติ”.
เพื่อให้การให้คำปรึกษาทางพันธุกรรม เป็นไปอย่างเหมาะสม จำเป็นต้องเตรียมดังต่อไปนี้
การจัดทำแผนผังเครือญาติ (pedigree) : ซักประวัติครอบครัวอย่างน้อย 3 รุ่นและบันทึกเป็นแผนผังเครือญาติ การกระจายของผู้ป่วยในแนวตั้งและแนวนอนช่วยคาดเดารูปแบบการถ่ายทอดได้การคาดเดารูปแบบการถ่ายทอด : ใช้แผนผังเครือญาติและลักษณะอาการเพื่อประเมินว่าเป็นแบบเด่นบนออโตโซม แบบด้อยบนออโตโซม แบบเชื่อมโยงกับโครโมโซม X หรือแบบถ่ายทอดทางมารดาการอธิบายการตรวจยีนและการขอความยินยอมเป็นลายลักษณ์อักษร : ก่อนตรวจข้อมูลทางพันธุกรรมของผู้ป่วย จำเป็นต้องอธิบายความหมายและข้อจำกัดของการตรวจอย่างเพียงพอ และขอความยินยอมเป็นลายลักษณ์อักษรการทำให้ผลตรวจ DNA ไม่ระบุตัวตนและการจัดการข้อมูล : ปกป้องความเป็นส่วนตัวด้วยการทำให้ไม่ระบุตัวตนแบบเชื่อมโยงได้การกำหนดแนวทางรับมือผลตรวจที่พบโดยบังเอิญล่วงหน้า : ยืนยันกับผู้ป่วยก่อนตรวจว่าจะมีการแจ้งอย่างไรหากพบผลตรวจที่พบโดยบังเอิญซึ่งรุนแรงและอาจเป็นอันตรายถึงชีวิต
ชนิดการตรวจ เป้าหมาย ลักษณะ วิธี PCR + การหาลำดับเบสแบบแซงเกอร์ การค้นหาการกลายพันธุ์ของยีนเดี่ยว ความแม่นยำสูง เหมาะสำหรับยืนยันตำแหน่งการกลายพันธุ์ที่ทราบแล้ว การตรวจแบบแผง (targeted sequencing) กลุ่มยีนที่เกี่ยวข้องกับโรคเฉพาะ ตรวจยีนที่เกี่ยวข้องกับโรคจอประสาทตา พร้อมกัน มีอัตราการวินิจฉัยสูง5) การวิเคราะห์เอ็กโซม (NGS) บริเวณเอ็กซอนทั้งหมด ตรวจพบความแปรผันที่ไม่ทราบมาก่อน มีประโยชน์เมื่อมียีนก่อโรคจำนวนมาก การวิเคราะห์จีโนมทั้งชุด (WGS) จีโนมทั้งหมด อาจมีอัตราการวินิจฉัยสูงกว่าการตรวจ NGS แบบมุ่งเป้าที่มีอยู่13)
ในโรคจอประสาทตา เสื่อมจากพันธุกรรม เมื่อสงสัยว่าเป็นโรคที่เกิดจากการกลายพันธุ์ของยีน RPE65 และในกรณีเช่นเพื่อช่วยพิจารณาความเหมาะสมสำหรับการรักษาด้วยยีน การตรวจบางรายการที่เข้าเกณฑ์สามารถดำเนินการเป็นการรักษาที่อยู่ในสิทธิ์ประกันได้4) .
การตีความความแปรผันต้องใช้ดุลยพินิจอย่างรอบคอบ เป็นที่ทราบกันว่าประมาณ 30% ของข้อความที่ตีพิมพ์ว่าเป็น “ความแปรผัน” ในบทความนั้นแท้จริงแล้วเป็นโพลีมอร์ฟิซึม (ความแปรผันปกติ) โดยทั่วไป การเปลี่ยนแปลงลำดับเบสที่พบในคนสุขภาพดีตั้งแต่ 1 คนขึ้นไปในทุก 100 คน ควรจัดเป็นโพลีมอร์ฟิซึม
มีการใช้ฐานข้อมูลหลักดังต่อไปนี้
OMIM (Online Mendelian Inheritance in Man) : ฐานข้อมูลแบบครอบคลุมของโรคและยีนทางพันธุกรรมGeneReviews : ให้ข้อมูลการปรึกษาทางพันธุกรรมสำหรับแต่ละโรคRetNet (Retinal Information Network) : ฐานข้อมูลยีนที่เชี่ยวชาญด้านโรคจอประสาทตา
Q
ค่าตรวจพันธุกรรมเบิกประกันได้หรือไม่?
A
ในโรคตาทางพันธุกรรม บางชนิด สามารถรับการตรวจทางพันธุกรรมเป็นการรักษาที่อยู่ในสิทธิ์ประกันได้เมื่อมีการพิจารณาข้อบ่งชี้ของการรักษาและเป็นไปตามเกณฑ์ของสถานพยาบาลที่กำหนด อย่างไรก็ตาม การครอบคลุมของประกันจะแตกต่างกันตามชนิดของการตรวจและโรคที่ตรวจ จึงต้องสอบถามที่สถานพยาบาลที่ไปตรวจ การตรวจที่ไม่อยู่ในสิทธิ์ประกันอาจต้องชำระเอง สำหรับโรคที่กำหนดเป็นโรคยากต่อการรักษา (เช่น retinitis pigmentosa) อาจได้รับการสนับสนุนทางการเงินผ่านระบบเงินช่วยเหลือค่ารักษาพยาบาลสำหรับโรคที่กำหนดเป็นโรคยากต่อการรักษาได้เช่นกัน4) .
ในการให้คำปรึกษาทางพันธุกรรม อุดมคติคือการทำงานร่วมกับแพทย์ผู้เชี่ยวชาญด้านพันธุศาสตร์และนักให้คำปรึกษาทางพันธุกรรมที่ได้รับการรับรอง ในโรงพยาบาลมหาวิทยาลัย อาจจำเป็นต้องผ่านการพิจารณาของคณะกรรมการจริยธรรม เนื่องจากข้อมูลทางพันธุกรรมอาจส่งผลไม่เพียงต่อผู้ป่วยเท่านั้น แต่ยังต่อสมาชิกในครอบครัวด้วย จึงต้องใช้ความระมัดระวังเป็นพิเศษในการจัดการข้อมูล นอกจากนี้ การอภิปรายทางสังคมเกี่ยวกับการป้องกันการเลือกปฏิบัติที่ไม่เป็นธรรมบนพื้นฐานของข้อมูลทางพันธุกรรมก็ยังคงดำเนินต่อไป8) .
โรคตาทางพันธุกรรม ต่อไปนี้ได้รับการกำหนดให้เป็นโรคหายาก และอยู่ในเกณฑ์ได้รับการช่วยเหลือค่าใช้จ่ายทางการแพทย์
เรตินิติสพิกเมนโตซา
ต้อหิน แต่กำเนิดกระจกตา ดิสโทรฟีแบบหยดเจลาติน
ผู้ป่วยโรคหายากจะได้รับการลดภาระค่าใช้จ่ายทางการแพทย์ส่วนที่ต้องจ่ายเอง มีการกำหนดเพดานรายเดือนสำหรับค่ารักษานอกโรงพยาบาล ค่ารักษาในโรงพยาบาล และค่ายาเมื่อรวมกัน และมีการใช้เกณฑ์ตามระดับรายได้
ยีนบำบัด voretigene neparvovec สำหรับโรคจอประสาทตา เสื่อมทางพันธุกรรมที่เกิดจากการกลายพันธุ์ของยีน RPE65 เป็นผลิตภัณฑ์ที่ให้ยาฉีดใต้จอประสาทตา โดยใช้เวกเตอร์ AAV2 ประสิทธิผลและความปลอดภัยได้รับการยืนยันแล้วในการทดลองแบบสุ่มมีกลุ่มควบคุม6) การตัดสินใจว่าการรักษาเหมาะสมหรือไม่ต้องยืนยันชนิดของโรคด้วยการตรวจยีน
สำหรับการรักษาด้วยแอนติเซนส์โอลิโกนิวคลีโอไทด์ (ASO) กำลังมีการศึกษาการฉีดเข้าวุ้นตา ของผลิตภัณฑ์สำหรับโรค Leber congenital amaurosis ชนิดที่ 10 (LCA10) ที่เกิดจากการกลายพันธุ์ของ CEP290 ในการทดลองทางคลินิก9) .
การปลูกถ่ายเซลล์เยื่อบุสีของจอประสาทตา แบบอัตโนมัติโดยใช้เซลล์ iPS กำลังได้รับการศึกษาในฐานะเวชศาสตร์ฟื้นฟูสำหรับโรคจุดภาพชัด เสื่อมตามอายุ7) แม้จะเป็นคนละสาขากับโรคจอประสาทตา เสื่อมทางพันธุกรรม แต่ก็ได้รับความสนใจในฐานะตัวอย่างจริงของการบำบัดด้วยเซลล์ของจอประสาทตา
การปรึกษาทางพันธุกรรมสามารถรับได้ที่โรงพยาบาลมหาวิทยาลัยและโรงพยาบาลที่มีแผนกเวชพันธุศาสตร์ทั่วประเทศ ต่อไปนี้เป็นข้อมูลอ้างอิง
สมาคมให้คำปรึกษาทางพันธุกรรมแห่งญี่ปุ่นเผยแพร่ข้อมูลสถานพยาบาลที่มีนักให้คำปรึกษาทางพันธุกรรมที่ได้รับการรับรอง
ศูนย์ข้อมูลโรคยาก Nanbyo ให้ข้อมูลจุดรับคำปรึกษาเกี่ยวกับโรคที่กำหนดและรักษายาก
ยังสามารถปรึกษาได้ที่แผนกเวชพันธุศาสตร์และคลินิกจักษุของโรงพยาบาลมหาวิทยาลัยที่มีจักษุแพทย์เฉพาะทาง
Q
ตอนนี้มีการบำบัดด้วยยีนแล้วหรือยัง?
A
ในโรคจอตาเสื่อมทางพันธุกรรมจากการกลายพันธุ์ของ RPE65 ประสิทธิผลและความปลอดภัยของ voretigene neparvovec ได้รับการแสดงให้เห็นใน RCT6) . เพื่อยืนยันข้อบ่งชี้ในการรักษา ต้องยืนยันชนิดของโรคด้วยการตรวจยีน สำหรับ LCA10 ที่มีการกลายพันธุ์ของ CEP290 กำลังศึกษาการฉีด ASO formulation เข้าวุ้นตา ในงานวิจัยทางคลินิก9) .
การถ่ายทอดแบบยีนเด่นบนออโตโซม ยีนด้อยบนออโตโซม และแบบเชื่อมโยงกับโครโมโซม X เป็นไปตามกฎของเมนเดล อย่างไรก็ตาม ปัจจัยต่อไปนี้อาจทำให้การคาดการณ์แบบง่ายทำได้ยาก
การแสดงออกไม่ครบถ้วน (penetrance) : แม้จะมียีนกลายพันธุ์ ก็ไม่ใช่ทุกคนที่จะเกิดโรค เมื่อ penetrance ต่ำ ลักษณะอาจข้ามรุ่น ทำให้ยากที่จะอนุมานรูปแบบการถ่ายทอดจากผังเครือญาติความแปรผันของการแสดงออก (expressivity) : แม้ในสมาชิกครอบครัวที่มียีนกลายพันธุ์เดียวกัน ความรุนแรงของอาการก็อาจแตกต่างกันการกลายพันธุ์แบบเพิ่มหน้าที่ (gain-of-function) : กลไกที่โปรตีนกลายพันธุ์ได้รับหน้าที่ที่เป็นอันตรายใหม่ ซึ่งแตกต่างจากผล dominant-negative แบบปกติ
ไมโทคอนเดรียอยู่ในไซโทพลาซึม และมีเพียง mtDNA ที่มาจากไข่ของมารดาเท่านั้นที่ส่งต่อไปยังบุตร ในแต่ละเซลล์มี mtDNA หลายพันสำเนา และอาจเกิดภาวะที่ mtDNA กลายพันธุ์กับ mtDNA ปกติปะปนกัน (heteroplasmy) ได้ ยิ่งสัดส่วนของ heteroplasmy สูง อาการมักยิ่งรุนแรง ใน Leber hereditary optic neuropathy (LHON ) การกลายพันธุ์ 3 ตำแหน่ง ได้แก่ 11778 (พบบ่อยที่สุด), 3460 และ 14484 คิดเป็นประมาณ 90% ของการกลายพันธุ์ทั้งหมด
ภาวะไดโซมีจากพ่อหรือแม่ฝ่ายเดียว คือภาวะที่โครโมโซมคู่หนึ่งทั้งสองแท่งมาจากพ่อหรือแม่คนเดียวกัน โดยไม่มีโครโมโซมจากอีกฝ่ายหนึ่ง เด็กที่เกิดจากผู้พาหะของโรคยีนด้อยแบบออโตโซมอาจป่วยได้แม้อีกฝ่ายจะไม่ใช่ผู้พาหะ ทำให้ดูเหมือนเป็นกรณีเกิดขึ้นเป็นครั้งคราว10) เมื่อรวมกับการกลายพันธุ์แบบ de novo และ heterozygote แบบผสม ควรพิจารณาโรคทางพันธุกรรมแม้ไม่มีประวัติครอบครัว
ใน LCA10 ที่มีการกลายพันธุ์ของ CEP290 มีการศึกษาการแก้ไขการสไปลซิงด้วยการรักษา ASO ในการทดลองทางคลินิก9)
ผู้สมัครการรักษาแบบตัดต่อยีน EDIT-101 ที่ใช้ CRISPR/Cas9 มีรายงานการพัฒนาในระยะก่อนคลินิกสำหรับ LCA10 (การกลายพันธุ์ของ CEP290)11)
การตรวจพันธุกรรมก่อนการฝังตัว (PGT-M) สามารถทำได้สำหรับโรคทางพันธุกรรมแบบเด่นและแบบด้อยบนออโตโซม และอาจพิจารณาได้ภายใต้กรอบจริยธรรม12)
การหาลำดับจีโนมทั้งชุด (WGS) มีแนวโน้มช่วยเพิ่มอัตราการวินิจฉัยระดับโมเลกุลในโรคจอประสาทตา ทางพันธุกรรม เมื่อเทียบกับการตรวจพันธุกรรมมาตรฐานที่มีอยู่13)
กำลังพัฒนาเครื่องมือคาดการณ์ความเป็นสาเหตุของความผิดปกติของยีนโดยใช้ปัญญาประดิษฐ์ (AI) และคาดว่าจะช่วยเพิ่มความแม่นยำในการตีความการกลายพันธุ์
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Sysmex. 遺伝性網膜ジストロフィの遺伝学的検査. URL: https://www.sysmex.co.jp/patients/ird_panel/feature/
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
日本医学会. 医療における遺伝学的検査・診断に関するガイドライン. 2022年3月改定. URL: https://jams.med.or.jp/guideline/genetics-diagnosis_2022.pdf
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
ถาม AI เกี่ยวกับบทความนี้
คัดลอกข้อความบทความแล้ววางในผู้ช่วย AI ที่คุณต้องการใช้
เปิดผู้ช่วย AI ด้านล่าง แล้ววางข้อความที่คัดลอกลงในช่องแชต