유전 상담은 ‘올바른 유전 정보를 제공하는 것’을 목적으로 하는 의료 서비스이다. 환자와 가족에게 유전 질환의 진단, 유전 양식, 재발 위험, 이용 가능한 검사, 치료법에 대한 정보를 제공하고, 스스로 결정할 수 있도록 돕는다. 이 정의는 국제적으로도 공유되며, 유전 상담의 세 가지 축은 ‘정보 제공’, ‘심리적 지원’, ‘의사결정 지원’3)이다.
유전성 안질환은 선천성 시각장애의 약 43%를 차지한다1). 자녀에서 염색체 이상이 나타나는 빈도는 약 0.51%이다. 적녹색각 이상은 남성 약 5%, 여성 약 0.2%에서 나타나는 가장 흔한 유전성 안질환 중 하나이다. 망막색소변성의 유병률은 약 1/4,0001/5,000으로, 시각장애의 주요 원인 질환 중 하나이다2).
유전 상담의 대상은 유전성 질환 환자 본인뿐 아니라, 발병 가능성이 있는 가족과 앞으로의 자녀에게 유전될 것을 걱정하는 보인자도 포함된다. 안과 의사는 진단을 맡는 동시에 유전 전문의와 인증 유전상담사와 협력하여 정보를 제공하는 역할을 한다.
Q유전 상담은 어디에서 받을 수 있나요?
A
대학병원이나 중핵병원의 유전자 진료 부문, 또는 인증 유전상담사가 근무하는 시설에서 받을 수 있다. 일본 유전상담학회와 일본인류유전학회의 인증 유전상담사 명단을 참고하면 시설을 찾을 수 있다. 난병정보센터 웹사이트에서도 상담처 안내를 제공한다.
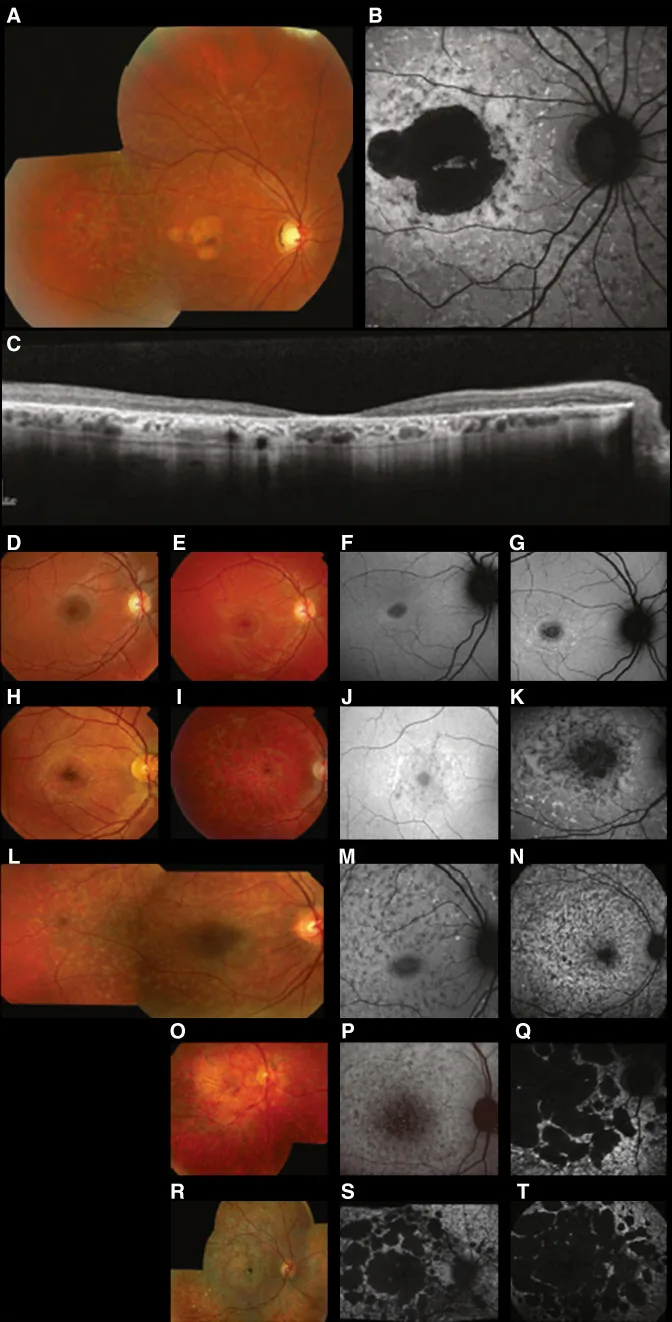
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
스타가르트병(STGD1)의 전형적인 다중모달 영상: 컬러 안저 사진에서는 망막색소상피 수준의 황백색 반점과 황반 위축이 보이고(A), 안저 자가형광에서는 황반의 저형광 영역과 주변의 비정상 형광이 보이며(B), SD-OCT에서는 외망막층과 RPE의 현저한 소실 및 반점에 대응하는 고반사 병소가 보인다(C). 이는 본문 “2. 유전 양식과 주요 유전성 안질환”에서 다루는 스타가르트병(ABCA4 유전자 변이에 의한 상염색체 열성 망막 이영양증)에 해당한다.
유전 방식에 따라 다릅니다. 상염색체 우성 유전에서는 영향을 받은 부모로부터 자녀에게 전달될 확률이 50%입니다. 상염색체 열성 유전에서는 부모가 모두 보인자일 때 자녀가 발병할 확률이 25%입니다. X연관 유전에서는 보인자인 어머니의 아들이 발병할 확률이 50%이며, 모계 유전(미토콘드리아)에서는 어머니가 환자이거나 보인자인 경우 모든 자녀에게 전달될 수 있습니다. 개별 상황은 유전 전문의 또는 공인 유전상담사와 상담하는 것이 권장됩니다.
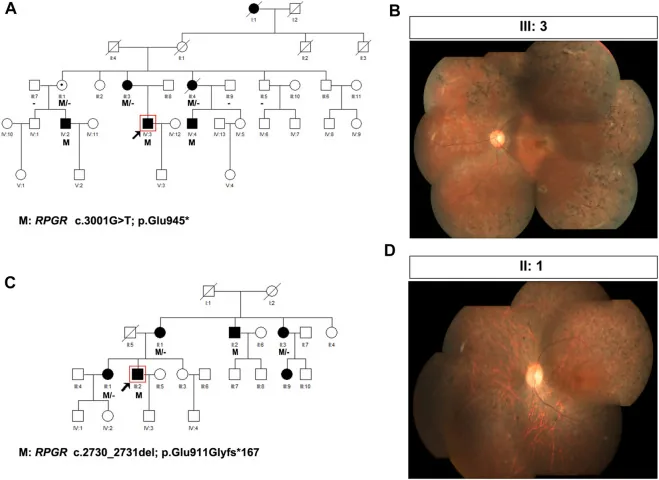
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
X-연관 망막색소변성(RPGR 변이: c.3001G>T 및 c.2730_2731del)을 가진 두 가계의 가계도와 안저 사진. 가계도는 표준 기호(채워진 사각형은 영향을 받은 남성, 점선 원은 보인자 여성을 의미)를 사용해 유전 양식을 보여 주며, 대응하는 안저 사진은 진행된 골침상 색소침착과 망막 위축을 보인다. 이는 본문 ‘4. 유전 상담의 실제와 유전자 검사’에서 다루는 가계도(pedigree) 작성과 유전 양식 판정에 해당한다.
변이를 해석할 때는 신중한 판단이 필요하다. 논문에 “변이”로 실린 설명의 약 30%는 다형성(정상 변이)인 것으로 알려져 있다. 일반적으로 정상인의 약 100명 중 1명 이상에서 나타나는 염기서열 변화는 다형성으로 보아야 한다.
주요 데이터베이스로는 다음이 사용된다.
OMIM(Online Mendelian Inheritance in Man): 유전 질환과 유전자의 종합 데이터베이스
GeneReviews: 질환별 유전 상담 정보를 제공
RetNet(Retinal Information Network): 망막 질환에 특화된 유전자 데이터베이스
Q유전자 검사 비용은 보험 적용이 되나요?
A
일부 유전성 안질환에서는 치료 적응 여부를 판단하거나 지정된 시설 기준을 충족하는 경우, 보험 진료로 유전학적 검사를 받을 수 있다. 다만 검사 종류와 대상 질환에 따라 적용 여부가 다르므로, 진료받는 기관에서 확인이 필요하다. 보험 적용이 되지 않는 검사는 본인 부담 진료가 될 수 있다. 난치병 지정 질환(망막색소변성증 등)에서는 난치병 의료비 지원제도를 통한 경제적 지원도 이용할 수 있을 가능성이 있다4).
유전 상담을 시행할 때는 유전 전문의와 공인 유전 상담사와의 협력이 이상적이다. 대학병원에서는 윤리위원회 심사가 필요할 수 있다. 유전 정보는 환자 본인뿐 아니라 가족에게도 영향을 미칠 수 있으므로, 정보 취급에 특별한 배려가 필요하다. 유전 정보에 근거한 부당한 차별을 막기 위한 사회적 논의도 진행되고 있다8).
RPE65 유전자 변이에 의한 유전성 망막이영양증에 대한 유전자 치료제 voretigene neparvovec은 AAV2 벡터를 사용한 망막하 투여 제제이다. 무작위 대조시험에서 유효성과 안전성이 확인되었다6). 치료 적응증을 판단하려면 유전자 검사로 병형을 확정해야 한다.
미토콘드리아는 세포질에 존재하며, 어머니의 난자에서 유래한 mtDNA만 자녀에게 전달된다. mtDNA는 한 세포당 수천 개의 사본이 존재하며, 변이 mtDNA와 정상 mtDNA가 함께 존재하는 상태(헤테로플라스미, heteroplasmy)가 생길 수 있다. 헤테로플라스미의 비율이 높을수록 증상은 더 심한 경향이 있다. Leber 유전 시신경병증(LHON)에서는 11778번(가장 많음), 3460번, 14484번의 3가지 변이가 전체 변이의 약 90%를 차지한다.
단친성 이배체는 한 쌍의 염색체를 같은 부모에게서 모두 받고, 다른 한쪽 부모의 염색체는 갖지 않는 상태를 말한다. 상염색체 열성 질환의 보인자에게서 태어난 아이는 다른 쪽 부모가 보인자가 아니더라도 발병할 수 있어, 겉보기에는 산발 사례처럼 보일 수 있다10). de novo 변이와 복합 이형접합체와 함께, 가족력이 없더라도 유전성 질환을 고려해야 한다.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.