El asesoramiento genético es un servicio médico cuyo objetivo es proporcionar información genética correcta. Ofrece a los pacientes y sus familias información sobre el diagnóstico, el patrón de herencia, el riesgo de recurrencia, las pruebas disponibles y las opciones de tratamiento de las enfermedades genéticas, y apoya la toma de decisiones autónoma. Esta definición también se comparte a nivel internacional, y los tres pilares del asesoramiento genético son la «provisión de información», el «apoyo psicológico» y el «apoyo para la toma de decisiones»3).
Las enfermedades oculares hereditarias representan aproximadamente el 43% de la discapacidad visual congénita1). La frecuencia de anomalías cromosómicas en la descendencia es de aproximadamente 0,5-1%. La discromatopsia rojo-verde es una de las enfermedades oculares hereditarias más comunes, y aparece en alrededor del 5% de los varones y del 0,2% de las mujeres. La prevalencia de la retinosis pigmentaria es de aproximadamente 1/4.000 a 1/5.000, y es una de las principales causas de discapacidad visual2).
El asesoramiento genético no solo está dirigido al propio paciente con una enfermedad hereditaria, sino también a los familiares que pueden desarrollarla y a los portadores preocupados por transmitirla a sus futuros hijos. El oftalmólogo, además de encargarse del diagnóstico, tiene la función de proporcionar información en colaboración con especialistas en genética y consejeros genéticos acreditados.
Q¿Dónde se puede recibir asesoramiento genético?
A
Se puede recibir en los departamentos de medicina genética de hospitales universitarios o hospitales de referencia, o en centros con consejeros genéticos acreditados. Se puede buscar un centro consultando los listados de consejeros genéticos acreditados de la Sociedad Japonesa de Asesoramiento Genético y la Sociedad Japonesa de Genética Humana. El sitio web del Centro de Información sobre Enfermedades Raras también ofrece orientación sobre dónde consultar.
2. Patrones de herencia y principales enfermedades oculares hereditarias
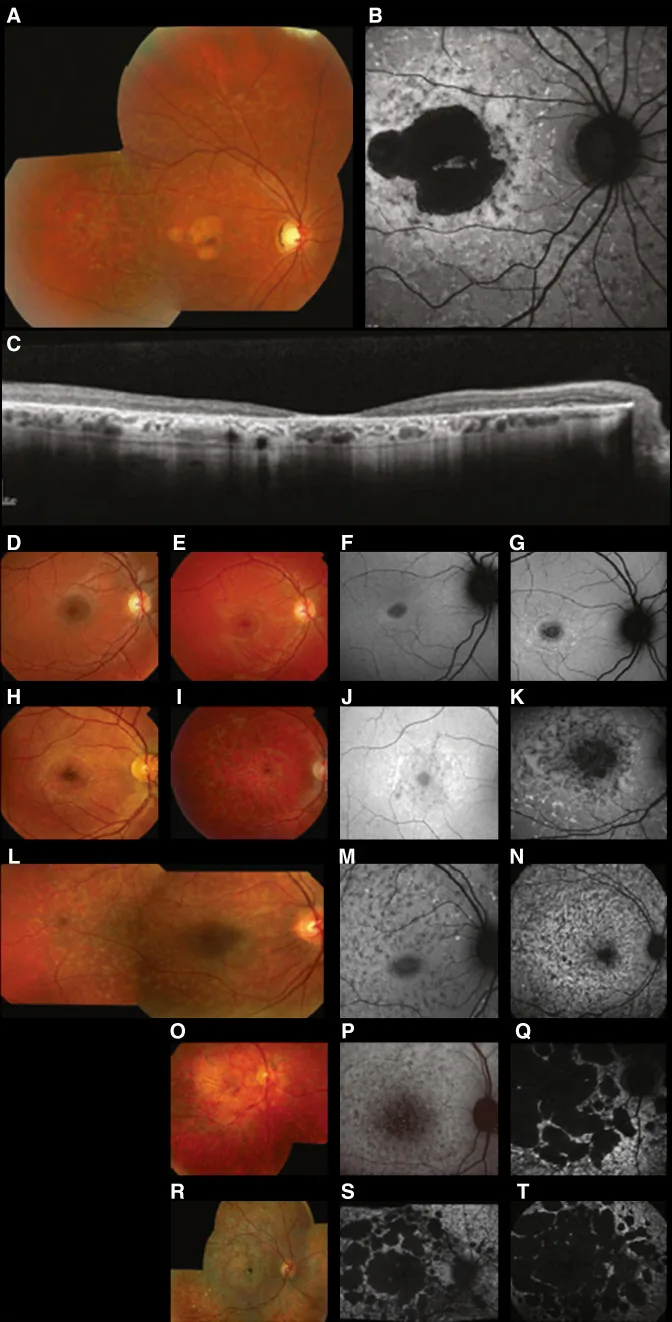
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Imágenes multimodales típicas de la enfermedad de Stargardt (STGD1): la fotografía de fondo de ojo en color muestra manchas amarillentas blanquecinas a nivel del epitelio pigmentario de la retina y atrofia macular (A); la autofluorescencia del fondo de ojo muestra una zona macular hipofluorescente con fluorescencia anormal alrededor (B); y la SD-OCT muestra una pérdida marcada de las capas retinianas externas y del EPR, con focos hiperrreflectivos que corresponden a las manchas (C). Corresponde a la enfermedad de Stargardt (una distrofia retiniana autosómica recesiva causada por mutaciones del gen ABCA4) tratada en la sección “2. Modos de herencia y principales enfermedades oculares hereditarias”.
El patrón de herencia es una información central en el asesoramiento genético y se clasifica en cuatro patrones principales.
Herencia autosómica dominante
Condición de aparición: La enfermedad aparece con una mutación en un alelo (estado heterocigoto).
Características del árbol genealógico: Los miembros afectados aparecen en generaciones consecutivas.
Riesgo de recurrencia: La probabilidad de transmitirla a un hijo es del 50%.
Nota: Si la penetrancia no es del 100%, puede saltarse una generación.
Herencia autosómica recesiva
Condición de aparición: La enfermedad aparece cuando ambos alelos están mutados (homocigoto o heterocigoto compuesto).
Características del árbol genealógico: Los miembros afectados aparecen entre hermanos. Los padres suelen ser portadores (heterocigotos).
Riesgo de recurrencia: La probabilidad de enfermedad en un hijo de dos portadores es del 25%.
Tendencia reciente: Como los matrimonios entre primos son menos frecuentes, ha aumentado la proporción de heterocigotos compuestos.
Herencia ligada al X
Condición de aparición: La mayoría de los pacientes son varones (hemicigotos).
En las mujeres: Como tienen dos cromosomas X, una mutación en uno de ellos las convierte en portadoras.
Características del árbol genealógico: Los afectados se concentran en varones y se transmite de la madre al hijo.
Riesgo de recurrencia: La probabilidad de que un hijo de una madre portadora desarrolle la enfermedad es del 50%.
Herencia materna (herencia mitocondrial)
Característica: El ADN mitocondrial (mtDNA) del espermatozoide se degrada casi por completo en la fecundación. Por eso solo se transmite de la madre al hijo.
QSi uno de los padres tiene una enfermedad ocular hereditaria, ¿cuál es la probabilidad de que se transmita al hijo?
A
El patrón de herencia varía. En la herencia autosómica dominante, la probabilidad de transmitirla de un progenitor afectado a un hijo es del 50%. En la herencia autosómica recesiva, si ambos padres son portadores, la probabilidad de que el hijo desarrolle la enfermedad es del 25%. En la herencia ligada al cromosoma X, la probabilidad de que un hijo varón de una madre portadora esté afectado es del 50%, y en la herencia materna (mitocondrial), si la madre está afectada o es portadora, puede transmitirse a todos los hijos. Para situaciones individuales, se recomienda consultar a un especialista en genética o a un consejero genético certificado.
3. Causas y factores de riesgo de las enfermedades oculares hereditarias
Los mecanismos de aparición de las enfermedades oculares hereditarias se clasifican según el tipo de efecto que una mutación genética tiene sobre la función de las proteínas.
Haploinsuficiencia (haploinsufficiency): El fenotipo aparece con la pérdida de la función de solo un alelo. Un ejemplo típico es la aniridia congénita (mutación de PAX6). Un alelo normal no puede aportar la cantidad de producto génico necesaria para la formación normal de los tejidos.
Efecto dominante negativo (dominant negative effect): La proteína mutante inhibe de forma competitiva y estructural la función de la proteína normal. En el síndrome de Marfan (mutación de FBN1), las moléculas mutantes de fibrilina-1 alteran la formación de la matriz extracelular.
Mutaciones mitocondriales: En la neuropatía óptica hereditaria de Leber (LHON), tres mutaciones puntuales—11778, 3460 y 14484—representan alrededor del 90% de todas las mutaciones. La alteración en la producción de energía daña las células ganglionares de la retina.
Mutaciones de novo: Mutaciones que no están presentes en los padres y surgen de nuevo durante la formación del óvulo o del espermatozoide. Un ejemplo representativo es la amaurosis congénita de Leber causada por mutaciones de CRX. También puede presentarse como casos aislados cuando nadie más de la familia está afectado.
Heterocigoto compuesto: En las enfermedades autosómicas recesivas, es una forma en la que se producen dos mutaciones diferentes en los dos alelos. Con la reciente disminución de los matrimonios entre primos, la proporción de heterocigotos compuestos ha aumentado en comparación con los homocigotos.
Disomía uniparental: Fenómeno en el que las dos copias del mismo cromosoma se heredan de un solo progenitor, mientras que falta el cromosoma del otro progenitor. Puede aparecer como un caso aparentemente esporádico10).
Se han identificado más de 100 genes causales de la retinosis pigmentaria, y un mismo fenotipo puede deberse incluso a muchos genes distintos2). Esta diversidad genética dificulta el diagnóstico.
4. Aspectos prácticos del asesoramiento genético y las pruebas genéticas
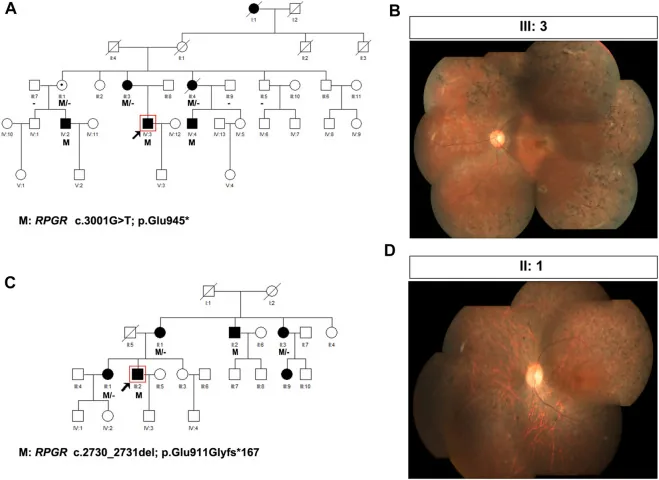
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Pedigríes y fotografías de fondo de ojo de dos familias con retinosis pigmentaria ligada al X (variantes de RPGR: c.3001G>T y c.2730_2731del). Los pedigríes usan símbolos estándar (cuadrados rellenos para varones afectados, círculos punteados para mujeres portadoras) para mostrar el patrón de herencia, y las fotografías de fondo de ojo correspondientes muestran pigmentación avanzada en espículas óseas y atrofia retiniana. Esto corresponde a la elaboración del pedigrí y la determinación del patrón de herencia que se tratan en la sección “4. Práctica del asesoramiento genético y las pruebas genéticas”.
Para realizar adecuadamente el asesoramiento genético, se necesita la siguiente preparación.
Creación de un pedigrí: Obtener antecedentes familiares de al menos tres generaciones y registrarlos como pedigrí. La distribución vertical y horizontal de las personas afectadas permite estimar el patrón de herencia.
Estimación del patrón de herencia: A partir del pedigrí y el fenotipo, inferir si es autosómico dominante, autosómico recesivo, ligado al X o materno.
Explicación de la prueba genética y obtención del consentimiento por escrito: Antes de examinar la información genética del paciente, es necesario explicar de forma suficiente el significado y las limitaciones de la prueba, y obtener el consentimiento por escrito.
Anonimización de los resultados de la prueba de ADN y gestión de la información: Proteger la privacidad mediante una anonimización que permita la vinculación.
Definición previa de la forma de actuar ante hallazgos incidentales: Confirmar con el paciente, antes de la prueba, la política de comunicación en caso de detectar hallazgos incidentales graves que puedan comprometer la vida.
Alta precisión. Adecuado para confirmar sitios de mutación conocidos
Prueba de panel (secuenciación dirigida)
Grupo de genes asociados con enfermedades específicas
Búsqueda simultánea de genes relacionados con enfermedades de la retina. Alta tasa de diagnóstico5)
Análisis del exoma (NGS)
Todas las regiones exónicas
Detecta variantes desconocidas. Es útil cuando hay muchos genes causantes de enfermedad
Análisis del genoma completo (WGS)
Genoma completo
Puede tener una tasa de diagnóstico más alta que las pruebas NGS dirigidas existentes13)
En la distrofia retiniana hereditaria, cuando se sospecha una enfermedad causada por una mutación del gen RPE65, y en casos como el de ayudar a determinar la idoneidad para la terapia génica, algunas pruebas que cumplen las condiciones pueden realizarse como atención cubierta por el seguro4).
Interpretación de las variantes y principales bases de datos
La interpretación de las variantes requiere una valoración cuidadosa. Se sabe que aproximadamente el 30 % de las descripciones publicadas como “variantes” en los artículos corresponden en realidad a polimorfismos (variantes normales). En general, los cambios en la secuencia que aparecen en 1 o más de cada 100 personas sanas deben considerarse polimorfismos.
Se utilizan las siguientes bases de datos principales.
OMIM (Online Mendelian Inheritance in Man): base de datos integral de enfermedades y genes hereditarios
GeneReviews: ofrece información de asesoramiento genético por enfermedad
RetNet (Retinal Information Network): base de datos genética especializada en enfermedades de la retina
Q¿El costo de las pruebas genéticas está cubierto por el seguro?
A
En algunas enfermedades oculares hereditarias, las pruebas genéticas pueden realizarse como atención cubierta por el seguro cuando se determina la indicación del tratamiento y se cumplen los criterios del centro designado. Sin embargo, la cobertura depende del tipo de prueba y de la enfermedad, por lo que es necesario confirmarlo en el centro al que se acuda. Las pruebas no cubiertas por el seguro pueden requerir pago de bolsillo. En las enfermedades designadas de difícil curación (como la retinitis pigmentosa), también puede haber apoyo económico mediante el programa de subsidio médico para enfermedades designadas de difícil curación4).
5. Sistema de asesoramiento genético y perspectivas del tratamiento
Al realizar asesoramiento genético, se considera ideal la colaboración con un especialista en genética y un asesor genético certificado. En los hospitales universitarios, puede requerirse la revisión de un comité de ética. Como la información genética puede afectar no solo al paciente sino también a su familia, se requiere especial cuidado en su manejo. También avanza el debate social sobre la prevención de la discriminación injusta basada en la información genética8).
Enfermedades raras designadas y ayuda para gastos médicos
En los pacientes con enfermedades raras, se reduce el copago médico. Se establece un tope mensual para la suma de consultas externas, hospitalización y dispensación de medicamentos, y se aplican categorías según los ingresos.
El fármaco de terapia génica voretigene neparvovec para la distrofia retiniana hereditaria causada por mutaciones del gen RPE65 es una preparación para administración subretiniana que utiliza un vector AAV2. Su eficacia y seguridad se han confirmado en ensayos aleatorizados controlados6). Para decidir si el tratamiento está indicado, primero debe confirmarse el tipo de enfermedad mediante pruebas genéticas.
En cuanto a la terapia con oligonucleótidos antisentido (ASO), se está estudiando en ensayos clínicos la administración intravítrea de una preparación para la amaurosis congénita de Leber tipo 10 (LCA10) causada por mutaciones en CEP2909).
El trasplante autólogo de células del epitelio pigmentario de la retina usando células iPS se está estudiando como medicina regenerativa para la degeneración macular asociada a la edad7). Aunque es un campo distinto de las enfermedades retinianas hereditarias, llama la atención como ejemplo real de terapia celular de la retina.
Q¿Ya está disponible la terapia génica?
A
En la distrofia retiniana hereditaria por mutación de RPE65, la eficacia y la seguridad de voretigene neparvovec se han demostrado en ensayos controlados aleatorizados6). Para confirmar si el tratamiento está indicado, es necesario definir el tipo de enfermedad mediante una prueba genética. En la LCA10 con mutación de CEP290, la inyección intravítrea de preparados de ASO se está estudiando en ensayos clínicos9).
La herencia autosómica dominante, autosómica recesiva y ligada al X sigue las leyes de Mendel. Sin embargo, los siguientes factores pueden dificultar una predicción simple.
Penetrancia: aunque una persona tenga la mutación, no todas desarrollan la enfermedad. Cuando la penetrancia es baja, el trastorno puede saltarse generaciones, lo que dificulta inferir el patrón de herencia a partir del árbol familiar.
Expresividad: incluso entre familiares con el mismo gen mutado, la gravedad de los síntomas puede variar.
Mutación de ganancia de función: mecanismo en el que la proteína mutada adquiere una nueva función dañina. Es diferente del efecto dominante negativo habitual.
Las mitocondrias se encuentran en el citoplasma, y solo el mtDNA procedente del óvulo de la madre se transmite al hijo. En cada célula hay miles de copias de mtDNA, y puede existir un estado en el que coexisten mtDNA mutado y mtDNA normal (heteroplasmia). Cuanto mayor es la proporción de heteroplasmia, más graves suelen ser los síntomas. En la neuropatía óptica hereditaria de Leber (LHON), tres variantes—11778 (la más frecuente), 3460 y 14484—representan aproximadamente el 90% de todas las variantes.
La disomía uniparental es una situación en la que ambos cromosomas de un par proceden del mismo progenitor y no hay cromosoma del otro progenitor. Un hijo de un portador de una enfermedad autosómica recesiva puede desarrollar la enfermedad aunque el otro progenitor no sea portador, dando lugar a un caso esporádico aparente10). Junto con las variantes de novo y los heterocigotos compuestos, debe considerarse una enfermedad hereditaria incluso cuando no haya antecedentes familiares.
7. Investigación más reciente y perspectivas futuras
En LCA10 con variantes de CEP290, la corrección del empalme mediante terapia ASO se está estudiando en ensayos clínicos9).
El candidato a tratamiento de edición génica EDIT-101, basado en CRISPR/Cas9, ha sido objeto de desarrollo preclínico para LCA10 (variantes de CEP290)11).
La prueba genética preimplantacional (PGT-M) puede realizarse para enfermedades hereditarias autosómicas dominantes y recesivas, y en algunos casos puede considerarse dentro de un marco ético12).
La secuenciación del genoma completo (WGS) ha mostrado que puede aumentar la tasa de diagnóstico molecular en las enfermedades retinianas hereditarias en comparación con las pruebas genéticas estándar existentes13).
Se están desarrollando herramientas de inteligencia artificial (IA) para predecir la patogenicidad de las variantes génicas, y se espera mejorar la precisión en la interpretación de variantes.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.