Genetic counseling is a medical service whose purpose is to provide accurate genetic information. It gives patients and families information about the diagnosis, inheritance pattern, recurrence risk, available tests, and treatment options for genetic diseases, and supports autonomous decision-making. This definition is shared internationally, and the three pillars of genetic counseling are ‘information provision’, ‘psychological support’, and ‘decision-making support’3).
Hereditary eye diseases account for about 43% of congenital visual impairment1). The frequency of chromosomal abnormalities in offspring is about 0.5-1%. Red-green color vision deficiency is one of the most common hereditary eye diseases, occurring in about 5% of males and 0.2% of females. The prevalence of retinitis pigmentosa is about 1 in 4,000 to 1 in 5,000, making it one of the major causes of visual impairment2).
The scope of genetic counseling includes not only the patient with the hereditary disease, but also family members who may develop it and carriers worried about passing it on to future children. Ophthalmologists play a role in diagnosis while working with genetic specialists and certified genetic counselors to provide information.
QWhere can genetic counseling be received?
A
It can be received in the genetics department of a university hospital or core hospital, or at a facility with certified genetic counselors. Facilities can be found by referring to the lists of certified genetic counselors from the Japan Society of Genetic Counseling and the Japan Society of Human Genetics. The website of the Nanbyo Information Center also provides guidance on where to consult.
2. Inheritance patterns and major hereditary eye diseases
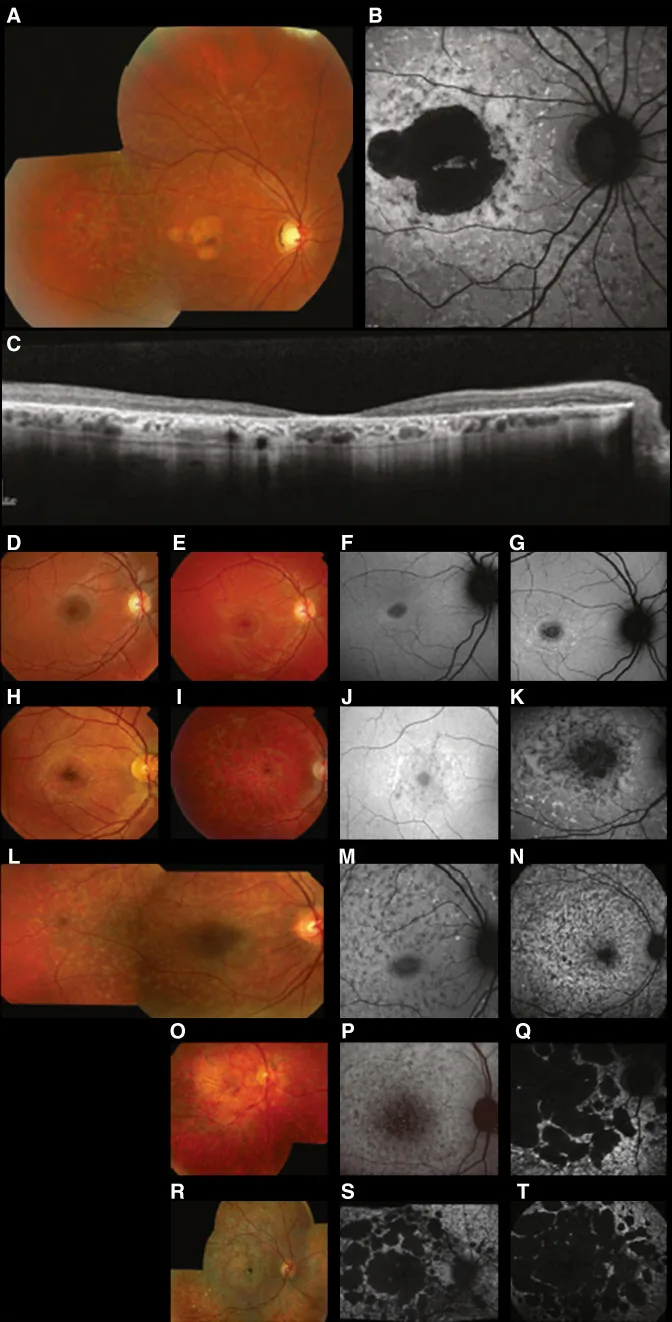
Fujinami K, et al. Br J Ophthalmol. 2024 Apr 8; 108(4):495-505. Figure 1. PMCID: PMC10958310. License: CC BY 4.0.
Typical multimodal images of Stargardt disease (STGD1): the color fundus photo shows yellow-white flecks at the level of the retinal pigment epithelium and macular atrophy (A); fundus autofluorescence shows a hypoautofluorescent area in the macula with abnormal fluorescence around it (B); and SD-OCT shows marked loss of the outer retinal layers and RPE, with hyperreflective foci corresponding to the flecks (C). This corresponds to Stargardt disease (an autosomal recessive retinal dystrophy caused by ABCA4 gene mutations) discussed in section “2. Inheritance Patterns and Major Hereditary Eye Diseases”.
The pattern of inheritance is core information in genetic counseling and is classified into four major patterns.
Autosomal dominant inheritance
Onset condition: The disease occurs with a mutation in one allele (heterozygous state).
Pedigree features: Affected members appear in successive generations.
Recurrence risk: The chance of passing it to a child is 50%.
Note: If penetrance is not 100%, a generation may be skipped.
Autosomal recessive inheritance
Onset condition: The disease occurs when both alleles are mutated (homozygous or compound heterozygous).
Pedigree features: Affected members appear among siblings. Parents are usually carriers (heterozygous).
Recurrence risk: The chance of disease in a child born to two carriers is 25%.
Recent trend: Because cousin marriages have become less common, the proportion of compound heterozygotes has increased.
X-linked inheritance
Affected individuals: Most patients are male (hemizygous).
Women: Because they have two X chromosomes, a mutation in one makes them a carrier.
Pedigree features: Affected people are predominantly male, and transmission is from mother to son.
Recurrence risk: The probability that a carrier mother’s son will be affected is 50%.
Maternal inheritance (mitochondrial inheritance)
Feature: Sperm mitochondrial DNA (mtDNA) is broken down almost completely at fertilization. Therefore it is passed only from the mother to the child.
QIf a parent has a hereditary eye disease, what is the chance it will be passed on to a child?
A
The inheritance pattern differs. In autosomal dominant inheritance, the chance of passing it from an affected parent to a child is 50%. In autosomal recessive inheritance, if both parents are carriers, the chance that a child will develop the disease is 25%. In X-linked inheritance, the chance that a son of a carrier mother will be affected is 50%, and in maternal inheritance (mitochondrial), if the mother is affected or a carrier, it may be passed to all children. For individual situations, consultation with a genetics specialist or certified genetic counselor is recommended.
3. Causes and risk factors for hereditary eye diseases
The onset mechanisms of hereditary eye diseases are classified by the type of effect a genetic mutation has on protein function.
Haploinsufficiency: A phenotype appears when the function of just one allele is lost. A classic example is congenital aniridia (PAX6 mutation). One normal allele cannot provide enough gene product for normal tissue formation.
Dominant negative effect: The mutant protein competitively and structurally inhibits the function of the normal protein. In Marfan syndrome (FBN1 mutation), mutant fibrillin-1 molecules disrupt formation of the extracellular matrix.
Mitochondrial mutations: In Leber hereditary optic neuropathy (LHON), three point mutations—11778, 3460, and 14484—account for about 90% of all mutations. Impaired energy production damages retinal ganglion cells.
De novo mutations: Mutations that are not present in the parents and arise newly during egg or sperm formation. A representative example is Leber congenital amaurosis caused by CRX mutations. They can also occur as isolated cases when no one else in the family tree is affected.
Compound heterozygote: In autosomal recessive disease, a form in which two different mutations occur in the two alleles. With the recent decline in cousin marriage, the proportion of compound heterozygotes has increased compared with homozygotes.
Uniparental disomy: A phenomenon in which both copies of the same chromosome are inherited from one parent, while the chromosome from the other parent is absent. It may appear as an apparently sporadic case10).
More than 100 causative genes for retinitis pigmentosa have been identified, and even the same phenotype can be caused by many different genes2). This genetic diversity makes diagnosis difficult.
4. Practical aspects of genetic counseling and genetic testing
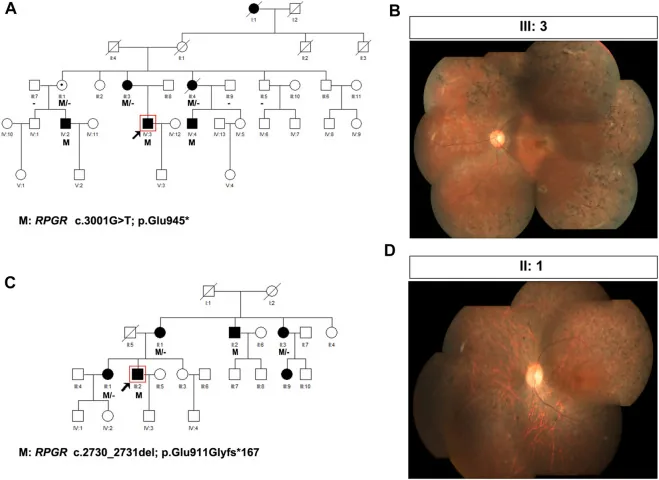
Liu X, Jia R, Meng X, et al. Analysis of RPGR gene mutations in 41 Chinese families affected by X-linked inherited retinal dystrophy. Front Genet. 2022;13:999695. Figure 1. PMID: 36276946; PMCID: PMC9582779; DOI: 10.3389/fgene.2022.999695. License: CC BY 4.0.
Pedigrees and fundus photographs of two families with X-linked retinitis pigmentosa (RPGR variants: c.3001G>T and c.2730_2731del). The pedigrees use standard symbols (filled squares for affected males, dotted circles for carrier females) to show the inheritance pattern, and the corresponding fundus photographs show advanced bone-spicule pigmentation and retinal atrophy. This corresponds to pedigree creation and inheritance pattern determination discussed in the section “4. Practical genetic counseling and genetic testing.”
The following preparation is needed to carry out genetic counseling appropriately.
Creating a pedigree: Obtain a family history covering at least three generations and record it as a pedigree. The vertical and horizontal distribution of affected people can help estimate the inheritance pattern.
Estimating the inheritance pattern: Use the pedigree and phenotype to infer whether it is autosomal dominant, autosomal recessive, X-linked, or mitochondrial.
Explaining genetic testing and obtaining written consent: Before examining a patient’s genetic information, it is necessary to provide a full explanation of the meaning and limitations of the test and obtain written consent.
Anonymizing DNA test results and managing information: Protect privacy through re-identifiable anonymization.
Deciding in advance how to handle incidental findings: Confirm with the patient before testing how to disclose any serious incidental findings that could affect life expectancy.
Highly accurate. Suitable for confirming known mutation sites
Panel testing (targeted sequencing)
A set of genes associated with specific diseases
Simultaneous search of genes related to retinal diseases. High diagnostic yield5)
Exome analysis (NGS)
All exonic regions
Detects unknown variants. Effective when there are many disease-causing genes
Whole-genome analysis (WGS)
Entire genome
May have a higher diagnostic yield than existing targeted NGS tests13)
In hereditary retinal dystrophy, when a disease caused by an RPE65 gene mutation is suspected, and in cases such as when the test is intended to help determine eligibility for gene therapy, some tests that meet the required conditions can be performed as insured medical care4).
Careful judgment is needed when interpreting variants. It is known that about 30% of descriptions published as “variants” in papers are polymorphisms (normal variants). In general, sequence changes that occur in one or more out of every 100 healthy individuals should be treated as polymorphisms.
The following major databases are used.
OMIM (Online Mendelian Inheritance in Man): A comprehensive database of genetic disorders and genes
GeneReviews: Provides genetic counseling information for each disease
RetNet (Retinal Information Network): A gene database specialized in retinal diseases
QIs the cost of genetic testing covered by insurance?
A
For some hereditary eye diseases, genetic testing can be provided as insured medical care when the treatment indication is being determined and the designated facility requirements are met. However, coverage depends on the type of test and the disease being tested, so confirmation at the facility you visit is necessary. Tests not covered by insurance may be paid for out of pocket. For designated intractable diseases (such as retinitis pigmentosa), financial support may also be available through the designated intractable disease medical expense subsidy program4).
5. Genetic counseling system and treatment prospects
When providing genetic counseling, collaboration with a genetics specialist and a certified genetic counselor is considered ideal. In university hospitals, review by an ethics committee may be required. Because genetic information can affect not only the patient but also family members, special care is needed in how it is handled. Social discussion is also progressing on preventing unfair discrimination based on genetic information8).
Designated intractable diseases and medical expense support
For patients with intractable diseases, out-of-pocket medical costs are reduced. A monthly cap is set for the combined total of outpatient care, hospitalization, and prescriptions, and income-based categories are applied.
The gene therapy drug voretigene neparvovec for inherited retinal dystrophy caused by RPE65 gene mutations is a subretinal formulation using an AAV2 vector. Its efficacy and safety have been confirmed in randomized controlled trials6). Deciding whether the treatment is indicated requires confirmation of the disease type through genetic testing.
As for antisense oligonucleotide (ASO) therapy, intravitreal administration of a formulation for Leber congenital amaurosis type 10 (LCA10) caused by CEP290 mutations is being studied in clinical trials9).
Autologous retinal pigment epithelium cell transplantation using iPS cells is being studied as regenerative medicine for age-related macular degeneration7). Although it is a different field from inherited retinal diseases, it is drawing attention as a real example of retinal cell therapy.
QIs gene therapy available now?
A
For hereditary retinal dystrophy caused by RPE65 mutations, the efficacy and safety of voretigene neparvovec have been shown in RCTs6). To confirm treatment eligibility, the disease subtype must be determined by genetic testing. In LCA10 with CEP290 mutations, intravitreal administration of ASO formulations is being studied in clinical trials9).
Autosomal dominant, autosomal recessive, and X-linked inheritance follow Mendel’s laws. However, the following factors can make simple prediction difficult.
Penetrance: Even if a person carries the mutation, not everyone develops the disease. When penetrance is low, the condition may skip generations, making it hard to infer the inheritance pattern from the family tree.
Expressivity: Even among family members with the same mutated gene, symptom severity can differ.
Gain-of-function mutation: A mechanism in which the mutated protein acquires a new harmful function. This differs from the usual dominant-negative effect.
Mitochondria are found in the cytoplasm, and only mtDNA from the mother’s egg is passed on to the child. Each cell contains thousands of copies of mtDNA, and a mixed state of mutant mtDNA and normal mtDNA (heteroplasmy) can occur. The higher the proportion of heteroplasmy, the more severe the symptoms tend to be. In Leber hereditary optic neuropathy (LHON), three variants—11778 (most common), 3460, and 14484—account for about 90% of all variants.
Uniparental disomy is a condition in which both chromosomes of a pair are inherited from the same parent, with no chromosome from the other parent. A child born to a carrier of an autosomal recessive disease may develop the disease even if the other parent is not a carrier, creating an apparent sporadic case10). Along with de novo variants and compound heterozygotes, hereditary disease should be considered even when there is no family history.
In LCA10 with CEP290 variants, splicing correction by ASO therapy is being studied in clinical trials9).
The genome-editing therapy candidate EDIT-101 using CRISPR/Cas9 has been reported in preclinical development for LCA10 (CEP290 variants)11).
Preimplantation genetic testing (PGT-M) can be performed for autosomal dominant and recessive hereditary diseases and may be considered under an ethical framework12).
Whole-genome sequencing (WGS) has been shown to potentially increase the molecular diagnostic rate compared with existing standard genetic tests for inherited retinal diseases13).
Artificial intelligence (AI)-based tools for predicting the pathogenicity of gene variants are being developed, and improved accuracy in variant interpretation is expected.
Solebo AL, Teoh L, Rahi J. Epidemiology of blindness in children. Arch Dis Child. 2017;102(9):853-857. doi:10.1136/archdischild-2016-310532.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795-1809.
National Society of Genetic Counselors’ Definition Task Force, Resta R, Biesecker BB, Bennett RL, Blum S, Hahn SE, Strecker MN, Williams JL. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns. 2006;15(2):77-83. doi:10.1007/s10897-005-9014-3. PMID:16761103.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, Taub DG, Janessian M, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(4):253-261. doi:10.1038/gim.2014.172. PMID:25412400; PMCID:PMC4572572.
Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet (London, England). 2017;390(10097):849-860. doi:10.1016/S0140-6736(17)31868-8. PMID:28712537; PMCID:PMC5726391.
Mandai M, Watanabe A, Kurimoto Y, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376(11):1038-1046. PMID: 28296613. doi:10.1056/NEJMoa1608368.
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature medicine. 2019;25(2):225-228. doi:10.1038/s41591-018-0295-0. PMID:30559420.
Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays : news and reviews in molecular, cellular and developmental biology. 2000;22(5):452-9. PMID:10797485.
Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nature medicine. 2019;25(2):229-233. doi:10.1038/s41591-018-0327-9. PMID:30664785.
De Wert G, Dondorp W, Shenfield F, Devroey P, Tarlatzis B, Barri P, Diedrich K, Provoost V, et al. ESHRE task force on ethics and Law22: preimplantation genetic diagnosis. Human reproduction (Oxford, England). 2014;29(8):1610-7. doi:10.1093/humrep/deu132. PMID:24927929.
Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology. 2016;123(5):1143-50. doi:10.1016/j.ophtha.2016.01.009. PMID:26872967; PMCID:PMC4845717.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.