Gelatinous drop-like corneal dystrophy (GDLD) is a hereditary corneal disease in which amyloid deposits beneath the corneal epithelium, causing significant bilateral visual loss.
This disease was first reported by Nakazumi in 1914, and since Kiyosawa named it “gelatinous drop-like corneal degeneration” in 1932, it has been called by this name. In the IC3D (International Committee for Classification of Corneal Dystrophies) classification, it is classified as an epithelial dystrophy with the abbreviation GDLD.
The causative gene is the TACSTD2 (tumor-associated calcium signal transducer 2) gene, identified by Tsujikawa et al. in 1999, which is a single-exon gene located on chromosome 1p324).
Prevalence: Rare worldwide, but reported to be relatively common in Japan 1). The incidence is thought to be decreasing due to a decline in consanguineous marriages 2)
Regional differences: Relatively common in Japan, rarely reported in Europe and the United States
Q118X mutation: A founder mutation in Japanese patients, accounting for over 80% of disease-causing chromosomes 2)
Age of onset: Often develops by the teenage years
In 2019, it was designated as a specified intractable disease (gelatinous drop-like corneal dystrophy) and became eligible for medical expense subsidies 2). Diagnostic criteria and severity classification have been established by the Ministry of Health, Labour and Welfare’s Research Project on Intractable Diseases 2).
QDoes GDLD occur outside Japan?
A
GDLD has been reported worldwide, but it is more common in Japan. There are almost no cases in Europe and the United States. More than 20 mutations in the TACSTD2 gene have been reported, indicating genetic heterogeneity. In Japan, the Q118X mutation on chromosome 1p32 is a frequent founder mutation, accounting for over 80% of disease-causing chromosomes in Japanese patients.
Often develops by the teenage years. The following symptoms are reported from early childhood.
Photophobia: A prominent symptom from the early stage
Foreign body sensation: Due to gelatinous elevations on the corneal surface
Lacrimation: Associated with irritative symptoms
Visual impairment: Gradually worsens with progression of amyloid deposition. Becomes marked after adulthood
The number and size of amyloid deposits increase with age. They become gray-white to yellow deposits, eventually covering most of the cornea, centered on the interpalpebral fissure 2). Vascular invasion from the periphery, marked visual loss, and ocular pain occur, and cosmetic issues also arise, significantly reducing the patient’s quality of life.
Clinical Findings (Findings Confirmed by Physician Examination)
Corneal opacities are classified into four types based on morphology. These can be distinguished by anterior segment observation using a slit-lamp microscope 2,3).
Mulberry-like
typical mulberry type: The most typical type.
Central cornea: Gray-white elevated lesions cluster together, resembling the appearance of a mulberry.
Subepithelial amyloid: Milky-white, translucent, gelatinous elevations increase from the center to the periphery.
Band-keratopathy type
band-keratopathy type: May be seen in the early stages.
Interpalpebral zone: Superficial opacities are observed. Findings resemble band keratopathy.
Conjunctival lesions: Lesions may also be present in the conjunctiva.
Kumquat-like
kumquat-like type: Common in advanced cases.
Diffuse yellowish-white deposits: The entire cornea turns yellow, resembling a kumquat.
Vascular invasion: May be accompanied by superficial neovascularization.
Stromal opacity type
Stromal opacity type: A more advanced stage.
Involvement of the stroma: The lesion extends to the corneal stroma.
Vascular invasion: Milky-white to yellow gelatinous elevations with vascular invasion.
Ide et al. reported a detailed clinical spectrum of 34 cases in Japan and showed that even with the same TACSTD2 gene mutation (Q118X homozygote), four phenotypes coexist3).
Other characteristic findings include the following:
Delayed fluorescein staining: Despite the absence of corneal epithelial damage, increased permeability due to tight junction dysfunction allows fluorescence to be observed a few minutes after fluorescein instillation2)
Epithelial thinning: The corneal epithelium is thinned over the gelatinous elevations
Vascular invasion: Superficial vascular invasion is observed in the peripheral cornea
Abnormalities in the TACSTD2 gene lead to loss of normal intracellular localization of Claudin 1 and Claudin 7, which are tight junction proteins in the corneal epithelium, resulting in impaired epithelial barrier function5). Consequently, proteins such as lactoferrin from tears invade the cornea, forming amyloid fibers that deposit beneath the epithelium. Nakatsukasa et al. molecularly demonstrated that TACSTD2 is essential for normal claudin localization in Japanese pedigrees, clarifying that the pathology of GDLD is due to tight junction dysfunction5).
Family history: Because it is an autosomal recessive disorder, there is a risk of developing the disease if both parents are carriers.
Japanese ancestry: The Q118X nonsense mutation accounts for over 80% of pathogenic chromosomes as a founder mutation in Japan 2)
Consanguineous marriage: Usually, the parents of the proband are consanguineous. However, compound heterozygotes from marriages between different families can also develop the disease.
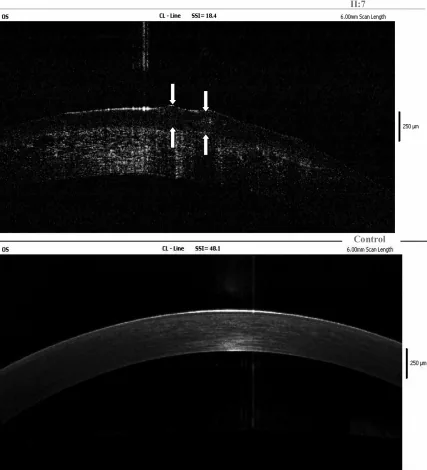
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Fourier-domain OCT findings of the left cornea of the proband show amyloid deposits in the corneal epithelium and superficial stroma, and arrows indicate the location of gelatinous drop-like lesions. This corresponds to the amyloid deposits discussed in the section “4. Diagnosis and testing methods.”
Diagnostic criteria (Ministry of Health, Labour and Welfare research group)
The diagnostic criteria for GDLD have been established by the Ministry of Health, Labour and Welfare’s Research Group on Policy Research for Intractable Diseases, “Research on the development and dissemination of clinical guidelines for anterior segment intractable diseases” 2). If a patient is judged as Definite according to these criteria, they become eligible for designated intractable diseases.
Gray-white elevated amyloid deposits (mulberry-like) are observed under the corneal epithelium in the central cornea to the palpebral fissure of both eyes.
Delayed staining is observed a few minutes after fluorescein staining despite the absence of corneal epithelial damage.
Superficial vascular invasion is observed in the peripheral cornea.
C. Differential diagnosis: Exclude secondary (acquired) corneal amyloidosis and climatic droplet keratopathy.
D. Extraocular complications: None.
E. Genetic testing: Abnormalities in the TACSTD2 gene are found.
Definite is met if any of the following conditions are satisfied2).
Cases that satisfy D, meet any of A, meet B1, and can exclude the diseases to be differentiated in C.
Cases that satisfy D, meet any of A, meet B2 or B3, meet E, and can exclude the diseases to be differentiated in C.
B1 (mulberry-like deposits) is a very characteristic finding, and diagnosis is not difficult in typical cases. In atypical cases, diagnosis is made by combining A to C with genetic testing (E)2).
Severity is classified into grades I to IV based on the best-corrected visual acuity in the better eye2).
Severity
Criteria
Medical Expense Subsidy
Grade I
Only one eye affected, the other eye is healthy
×
Grade II
Both eyes affected, best corrected visual acuity in the better eye ≥ 0.3
×
Grade III
Both eyes affected, best corrected visual acuity in the better eye ≥ 0.1 and < 0.3
○
Grade IV
Both eyes affected, best corrected visual acuity in the better eye < 0.1
○
A diagnosis of Definite qualifies for designated intractable diseases, and medical expense subsidies are available for severity Grade III or higher 2). If the better eye has visual field narrowing (central residual visual field ≤ 20 degrees with Goldmann I/4 target) due to secondary glaucoma, etc., the severity is upgraded by one level.
Slit-lamp microscopy: Observe gray-white elevated lesions from the central cornea to the palpebral fissure area, and differentiate into four types (mulberry-like, band-shaped, kumquat-like, and parenchymal opacity type).
Fluorescein penetration test (delayed staining): Due to tight junction dysfunction, the dye rapidly penetrates into the corneal tissue 2)
TACSTD2 genetic testing: Covered by insurance since fiscal year 2020 as corneal dystrophy genetic testing (D006-20) 2). TACSTD2 is a single-exon gene, making analysis easy. Particularly useful for diagnosing atypical cases
Histological examination (corneal section): Amyloid is confirmed by orange-red staining with Congo red and apple-green birefringence under polarized light microscopy
Secondary corneal amyloidosis: Amyloid deposits due to chronic irritation such as trichiasis, entropion, keratoconus protrusion, or hard contact lens wear. Differentiating features include no family history and a background of chronic ocular surface inflammation. May present with gelatinous droplike or lattice-like findings; histological examination is necessary for confirmation
Climatic droplet keratopathy: More common in men over 40 years old. Found in desert or extremely cold regions, caused by ultraviolet radiation and dryness. Presents with yellow to gray-white raised corneal lesions
Band keratopathy: Calcium salts deposit in the subepithelium. Begins at the periphery at 3 and 9 o’clock positions and progresses centrally
Lattice corneal dystrophy type I: Autosomal dominant inheritance due to TGFBI gene R124C mutation. Presents with branched filamentous opacities in the corneal stroma
TACSTD2 genetic testing has been covered by insurance since 2020 as “Corneal Dystrophy Genetic Testing (D006-20).” However, the facility must be certified and have the capability to perform the test. TACSTD2 is a single-exon gene, making analysis easy, and over 80% of Japanese patients have the Q118X founder mutation, making it particularly useful for diagnosing atypical cases 2).
Treatment for GDLD is selected based on the extent of opacities and the degree of visual impairment. Since it is a hereditary disease, the major challenge is the extremely high recurrence rate with any treatment 2). Many cases lead to blindness due to complications from multiple corneal transplants or secondary glaucoma.
Artificial tears: Used as symptomatic treatment to relieve surface irritation.
Continuous wear of therapeutic soft contact lenses (SCL): Can suppress recurrence of gelatinous elevated lesions and prolong the interval between surgeries.
Continuous wear of therapeutic SCL is considered as a conservative and adjunctive treatment 6). In 2020, Maeno et al. showed in a prospective observational study of GDLD patients that therapeutic SCL wear significantly suppressed recurrence of gray-white to yellow gelatinous elevated lesions 7). It is also recommended for postoperative recurrence prevention.
Purpose: To cover the ocular surface with graft-derived corneal epithelium and prevent re-invasion of host epithelium 10,11).
Multiple reports from Japan have described long-term outcomes of PTK. Oura et al. reported long-term results of PTK for GDLD cases, showing usefulness in prolonging the recurrence-free interval 8). Hieda et al. analyzed recurrence timing and clinical outcomes after PTK in a multicenter study in Japan 9).
Combined corneal limbal stem cell transplantation (LSCT) is internationally recognized as a Japanese-originated approach. Shimazaki et al. reported in 2002 the efficacy of LSCT-combined corneal transplantation for GDLD, showing that it can prolong the recurrence-free interval by suppressing re-invasion of host epithelial cells 10). Subsequently, Movahedan et al. also reported a similar approach 11).
To cover the ocular surface with graft-derived corneal epithelium, the host corneal epithelium is removed before performing limbal transplantation. After surgery, continuous wear of therapeutic SCL is continued to further delay recurrence.
In recent years, the indication of artificial cornea (Boston type I Kpro) has also been considered. Since it does not involve the host corneal epithelium, amyloid re-deposition can theoretically be avoided, but there are risks of postoperative complications such as infection and retroprosthetic membrane.
QDoes corneal transplantation still lead to recurrence?
A
In GDLD, the recurrence rate after corneal transplantation is extremely high. It has been reported that approximately 97% recur within 4 years after penetrating keratoplasty (PKP). The main cause is the replacement of graft epithelium by recipient epithelial cells. As a countermeasure originating in Japan, combined use of corneal limbal stem cell transplantation 10) and continuous wear of therapeutic SCL 7) is used to delay recurrence. In long-term management, the goal is to maintain visual function and prolong the interval between surgeries, assuming recurrence.
TACSTD2, the causative gene of GDLD, is a single-exon gene located on chromosome 1p32. In 1999, Tsujikawa et al. identified it as the causative gene through linkage analysis of Japanese families 4). The TACSTD2 protein plays an essential role in maintaining the barrier function of the corneal epithelium.
When a loss-of-function mutation occurs in the TACSTD2 gene, the normal intracellular localization of Claudin 1 and Claudin 7, which are tight junction constituent proteins, is impaired. Nakatsukasa et al. showed through analysis of cultured corneal epithelial cells and Japanese families that loss of TACSTD2 function leads to loss of Claudin localization at the apicolateral junction and reduced epithelial barrier function 5). Furthermore, in 2011, they reported novel TACSTD2 mutations in three families and their abnormal intracellular localization 12).
Due to reduced epithelial barrier function, proteins such as lactoferrin in tears invade the cornea. The invading lactoferrin forms amyloid fibers and deposits under the corneal epithelium. Although amyloid deposits contain lactoferrin, this disease is not due to an abnormality in the lactoferrin gene.
Histologically, the subepithelial milky opacities stain orange-red with Congo red and show apple-green birefringence under polarized light microscopy. Electron microscopy reveals that the epithelial tight junctions are replaced by electron-lucent spaces. Deposits also invade the corneal lamellae, causing degeneration of collagen fibers and proteoglycans.
More than 20 mutations in the TACSTD2 gene have been reported 12). In Japan, the Q118X mutation (nonsense mutation, functional null) accounts for over 80% of disease chromosomes as a founder mutation 2). Homozygotes typically develop the disease, but compound heterozygotes from intermarriage between different families can also be affected. Interestingly, even among individuals with the same Q118X homozygosity, four clinical subtypes (mulberry-like, band-shaped, kumquat-like, and stromal opacity type) are observed 3).
Corneal amyloidosis is classified as primary or secondary, and systemic or localized. GDLD, along with lattice corneal dystrophy, is classified as a primary localized amyloidosis. Secondary localized amyloid degeneration occurs with trichiasis, keratoconus, trauma, long-term contact lens wear, etc., and is a differential diagnosis.
GDLD is a rare disease, and there was a challenge that few physicians at individual institutions had clinical experience, and standard diagnostic and treatment methods were not established. The Ministry of Health, Labour and Welfare’s Research Project on Intractable Diseases, “Epidemiological Survey Research Group for Rare Intractable Corneal Diseases” and “Research Group for Development and Dissemination of Clinical Guidelines for Anterior Segment Intractable Diseases” formulated diagnostic criteria and severity classification 2). In 2019, it was designated as a specified intractable disease “Gelatinous Drop-like Corneal Dystrophy,” and a Minds (Medical Information Network Distribution Service)-compliant clinical practice guideline is currently under development 2).
Long-term issues include recurrence and multiple treatments. Combining therapeutic SCL and limbal transplantation may delay recurrence and prolong the interval between surgeries, potentially improving long-term prognosis 6, 7, 10).
Reports of Atypical Cases and Unilateral Recurrence
Maeno et al. reported a clinically atypical case showing recurrent amyloid deposition in only one eye, demonstrating the phenotypic diversity of GDLD 13). TACSTD2 genetic testing plays a decisive role in diagnosing such atypical cases 2).
In basic research, detailed elucidation of tight junction molecular dynamics downstream of TACSTD2 and development of therapies targeting Claudin stabilization are expected. Clinically, expansion of indications for regenerative medicine approaches such as artificial corneas, corneal epithelial sheet transplantation, and iPS cell-derived corneal epithelium is being considered.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.